


Comparative Study on Bacterial Diversity of Zha-Chili in Different Regions
-
摘要: 本研究使用Illumina MiSeq测序技术解析兴山和保靖鲊广椒细菌群落结构。α多样性分析表明,兴山鲊广椒群落多样性及丰度均低于保靖鲊广椒(P<0.05)。两地鲊广椒中细菌主要是隶属于厚壁菌门(Firmicutes)的Lactobacillus(乳酸杆菌属),其在兴山鲊广椒中含量为50.24%,在保靖鲊广椒中为88.26%。β多样性分析发现,兴山和保靖鲊广椒细菌群落结构整体上具有显著差异(P<0.01),同时发现11个差异显著的细菌类群,其中得分较高的为Lactobacillus和Chishuiella(赤水河菌属)。表型预测发现,兴山鲊广椒细菌类群在可移动原件、生物膜形成、革兰氏阴性、致病潜力和氧化胁迫耐受等表型上表达显著,而保靖鲊广椒细菌类群则在革兰氏阳性表型上表达显著。Abstract: In this study, Illumina MiSeq sequencing technology was used to analyze the bacterial community structure of Zha-Chili from Xingshan and Baojing. The α-diversity analysis showed that the community diversity and abundance of Zha-Chili in Xingshan were significantly lower than those in Baojing (P<0.05). Lactobacillus belonging to the Firmicutes was the main bacteria in the two places, the abundance of which was 50.24% in Xingshan and 88.26% in Baojing. β diversity analysis found that the bacterial community structure of Xingshan and Baojing Zha-Chili was significantly different on the whole (P<0.01) and 11 biomarkers were identified, among which Lactobacillus and Chishuiella were the most influential ones. Phenotypes prediction showed that bacterial groups of Zha-Chili of Xingshan were significantly expressed in movable elements, biofilm formation, gram negative, pathogenic potential and tolerance to oxidative stress, while the bacteria in Baojing Zha-Chili were significantly expressed in gram-positive.
-
鲊广椒是我国特色发酵食品中的一种,因其独特发酵风味而广泛流传在湖南和湖北等地区,其中拥有适宜地理位置和气候条件的兴山和保靖亦是鲊广椒生产和销售的重要区域。鲊广椒的制作工艺较为简单,主要是将玉米粉、辣椒、胡椒和花椒等原料均匀混合后进行密封发酵,发酵完成即为成品。由此可知鲊广椒在制作过程中,发酵过程至关重要。微生物作为发酵过程的主体,其种类和数量亦会对鲊广椒的品质产生影响[1]。目前已有不同地区鲊广椒细菌群落多样性的相关研究,通过研究宜昌鲊广椒的细菌多样性,王玉荣等[2]发现其优势细菌属为乳酸杆菌属、魏斯氏菌属和片球菌属等,其中乳酸杆菌属对风味的形成有促进作用。崔小丽等[3]对遵义鲊辣椒进行研究,结果发现鲊辣椒中主要菌属为乳酸菌、酵母菌和醋酸菌。李娜等[4]对咸丰和当阳的细菌群落结构进行了探究和比较,结果发现咸丰鲊广椒菌群丰度和多样性均显著高于当阳鲊广椒。由于不同地区鲊广椒的制作工艺和环境条件等因素均有所差异,因此其可能会对鲊广椒细菌群落结构产生影响[5-6]。综上所述,探究不同地区鲊广椒的细菌群落结构对鲊广椒品质提升具有重要意义。
MiSeq测序技术具有通量高、测序时间短和结果准确等特点,使全面和准确的揭示样品微生物群落结构成为现实[7]。随着Illumina MiSeq高通量测序技术的快速发展,其已成为微生物分析方法中常用技术之一,在食品领域中亦有应用,常用于豆瓣酱[8]、发酵辣椒[9]和泡菜[10]等传统发酵食品的研究中。兴山和保靖作为鲊广椒制作和销售的重要地区,其内亦有丰富的鲊广椒资源,为鲊广椒的研究提供了良好条件。本研究分别从湖北省兴山县和湖南省保靖县采集了农家自制的鲊广椒样品,使用Illumina MiSeq测序技术解析其细菌群落结构的基础上,结合多元统计学方法对其细菌类群进行统计分析,同时对关键细菌类群进行甄别。通过本研究的实施,为鲊广椒的品质提升和菌种资源挖掘提供一定的理论支撑。
1. 材料与方法
1.1 材料与仪器
鲊广椒样品 共采集19份(原料均为玉米碴),其中10份分别采集自湖北省宜昌市兴山县惠农菜场和永安农贸市场(编号为XS1~XS10),其余9份则采集自湖南省湘西土家族自治州保靖县的商贸中心菜市场(编号为BJ1~BJ9)。样品采集后使用自封袋进行封装,密封完成后置于盛有冰袋的采样箱中迅速运送回实验室;微生物基因提取试剂盒 德国QIAGEN公司;dNTPs Mix、FastPfu Fly DNA Polymerase、10×聚合酶链式反应缓冲液 北京全式金生物技术有限公司;正向引物338F(5'-ACTCCTACGGGAGGCAGCA-3')、反向引物806R(5'-GGACTACHVGGGTWTCTAAT-3')正反向引物 武汉天一辉远生物科技有限公司。
Illumina MiSeq高通量测序平台 美国Illumina公司;Vetiri梯度基因扩增仪 美国AB公司;ND-2000C微量紫外分光光度计 美国Nano Drop公司;R930机架式服务器 美国DELL公司;CR21N型高速离心机 日本日立金属株式会社;FluorChem FC3化学发光凝胶成像系统 美国ProteinSimple公司。
1.2 实验方法
1.2.1 鲊广椒DNA提取、序列扩增及高通量测序
根据DNA提取试剂盒的步骤对鲊广椒DNA进行提取,参照AGNIESZKA扩增方法和体系[11]进行DNA扩增,扩增完成后,将检测合格的产物送至上海美吉生物医药科技有限公司进行测序。
1.2.2 序列质控和生物信息学分析
参照郭壮等[12]方法对序列进行配对和质控,下机后的数据使用QIIME(v1.9.0)平台[13]对合格序列进行分析:a.序列比对:使用PyNAST软件[14]对序列进行排齐处理;b.分类操作单元的构建:选用UCLUST方法构建分类操作单元[15](Operational taxonomic units,OTU);c.使用ChimeraSlayer软件对含有嵌合体序列的OTU进行切除[16];d.选择合适的OTU序列与Greengenes、SILVA和Ribosomal database project数据库[17-19]进行比对,进而获得各分类学地位;e.通过计算样品的超1指数和香农指数进行α多样性分析;f.在计算UniFrac距离的基础上进行β多样性分析[20]。
1.2.3 Bugbase表型预测
相关数据和样品分类信息上传至Bugbase(https://bugbase.cs.umn.edu/)以完成表型预测分析[21]。
1.3 数据处理
使用Mann-Whitney检验对兴山和保靖鲊广椒细菌群落多样性指数进行差异分析;使用主坐标分析计算样品间的距离;使用LEfSe(Linear discriminant analysis effect size,线性判别分析)分析甄别对鲊广椒造成差异的关键细菌类群[22]。使用Origin 2017软件完成箱形图和柱状图的绘制,使用R软件对鲊广椒微生物群落结构和表型预测进行多样性指数的差异分析,同时完成小提琴图的绘制。
2. 结果与分析
2.1 鲊广椒样品的α多样性分析
α多样性指数常用来反映样本微生物群落多样性和丰度,本研究选用超1指数和香农指数对鲊广椒的细菌多样性进行了评价,结果如图1所示。
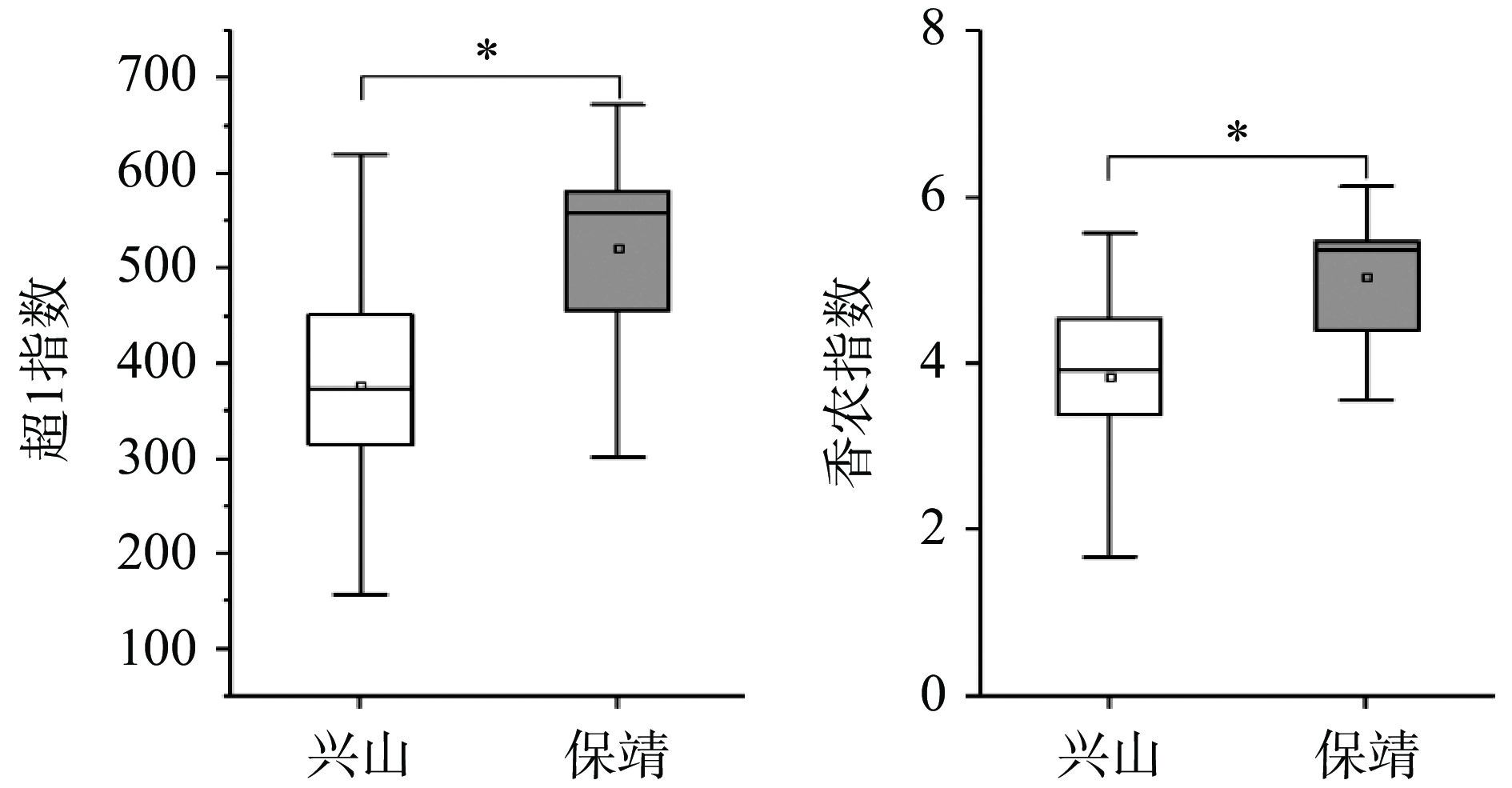
![]() 图 1 香农和超1指数的箱形图注:*表示差异显著(P<0.05),**表示差异非常显著(P<0.01),***表示差异极显著(P<0.001);图5同。Figure 1. Shannon and Chao 1 index box graph
图 1 香农和超1指数的箱形图注:*表示差异显著(P<0.05),**表示差异非常显著(P<0.01),***表示差异极显著(P<0.001);图5同。Figure 1. Shannon and Chao 1 index box graph由图1可知,兴山鲊广椒的超1和香农指数分别为377和3.83,而保靖鲊广椒的分别为521和5.05。经Mann-Whitney检验发现,两地区鲊广椒细菌群落的超1和香农指数均存在显著差异(P<0.05),由此说明兴山鲊广椒的细菌群落多样性与丰度均显著低于保靖鲊广椒。
2.2 鲊广椒微生物细菌群落的结构分析
本研究进一步在门和属水平上对鲊广椒样品进行了分析,同时将平均相对含量大于1%的细菌类群归为优势细菌门和优势细菌属。其中基于门水平的鲊广椒群落结构情况如图2所示。
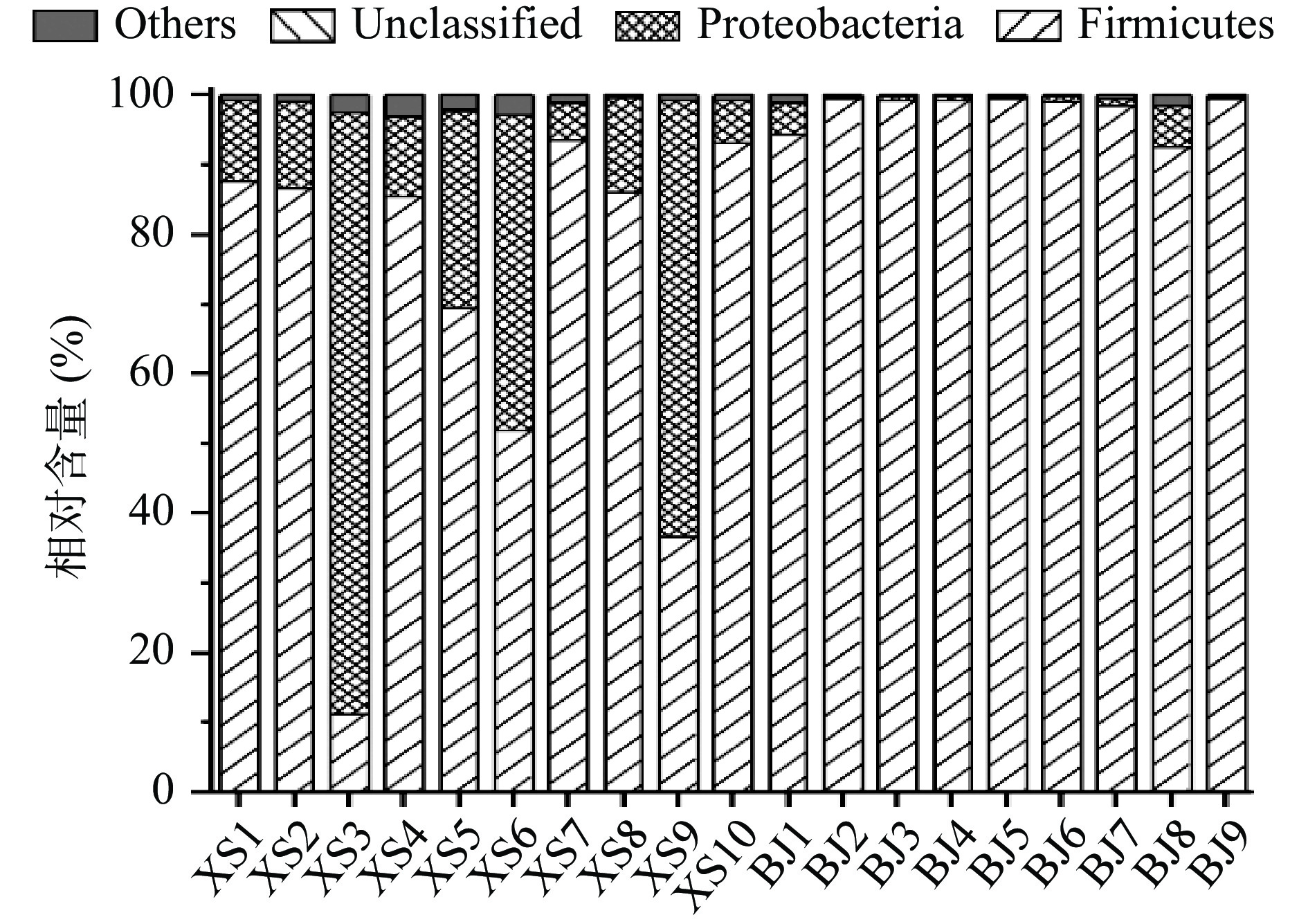
![]() 图 2 基于门水平的鲊广椒细菌多样性分析Figure 2. Analysis of bacterial diversity of Zha-Chili based on phylum level
图 2 基于门水平的鲊广椒细菌多样性分析Figure 2. Analysis of bacterial diversity of Zha-Chili based on phylum level由图2可知,鲊广椒样品中共发现16个细菌门,其中Firmicutes(厚壁菌门)和Proteobacteria(变形菌门)为兴山和保靖鲊广椒中共有的优势细菌门,二者在兴山鲊广椒中的平均相对含量分别为70.21%和28.28%,在保靖鲊广椒中则分别为97.98%和1.58%。经Mann-Whitney检验发现,兴山鲊广椒中Firmicutes和Proteobacteria的平均相对含量均显著低于保靖鲊广椒(P<0.001)。基于属水平的鲊广椒群落结构分析如图3所示。
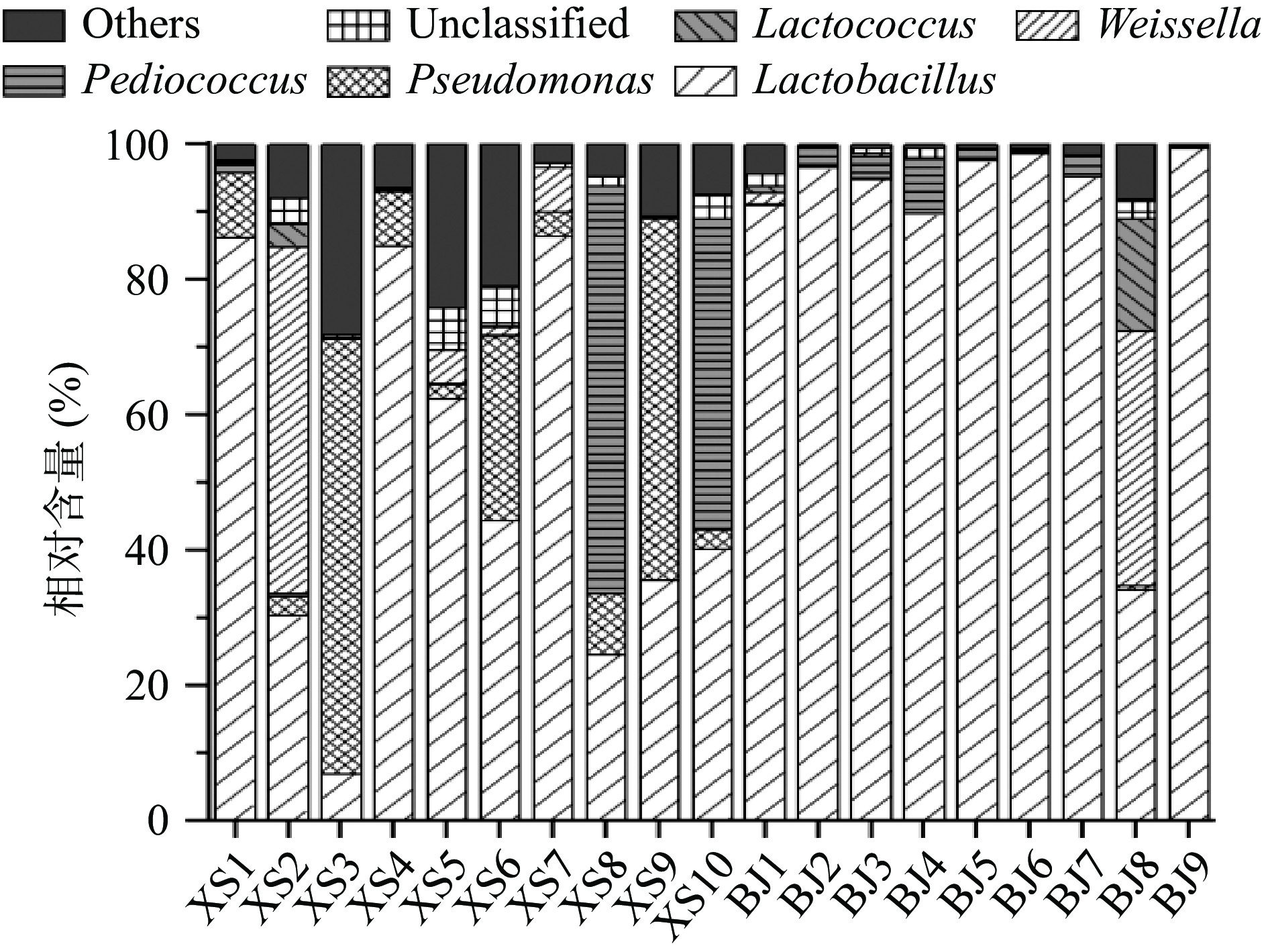
![]() 图 3 基于属水平的鲊广椒细菌多样性分析Figure 3. Bacterial diversity analysis of Zha-Chili based on genus level
图 3 基于属水平的鲊广椒细菌多样性分析Figure 3. Bacterial diversity analysis of Zha-Chili based on genus level由图3可知,鲊广椒样品中共发现279个细菌属,其中兴山鲊广椒中优势细菌属共有5个,分别为隶属于Firmicutes的Lactobacillus(乳酸杆菌属,50.24%)、Pediococcus(乳酸片球菌属,10.84%)和Weissella(魏斯氏菌属,6.44%),隶属于Proteobacteria的Pseudomonas(假单胞菌属,18.29%)和Serratia(沙雷氏菌属,1.57%)。保靖鲊广椒中优势细菌属共有4个,分别为隶属于Firmicutes的Lactobacillus(88.26%)、Weissella(4.40%)、Pediococcus(2.11%)和Lactococcus(乳球菌属,1.97%)。此外兴山和保靖鲊广椒中乳酸菌的含量均较高,在兴山中达67.52%,在保靖中则高达96.72%,说明鲊广椒在发酵过程中,乳酸菌占据主导地位。经Mann-Whitney检验发现,Lactobacillus在保靖鲊广椒中的平均相对含量显著高于兴山鲊广椒(P<0.05),而Pseudomonas则呈现相反趋势(P<0.05)。Lactobacillus是一类严格厌氧的革兰氏阳性、无芽孢形成的杆状或球杆菌,和Pediococcus、Weissella和Lactococcus一样,均为乳酸菌家族的一员,其在发酵肉制品、泡菜和发酵奶等发酵食品中广泛存在,可以利用食品中水溶性碳水化合物产生大量乳酸[23],在赋予食品独特发酵滋味的同时还能起到一定的延长保质期作用[24-26]。相较于Lactobacillus的厌氧,Pseudomonas则为一种需氧或兼性厌氧的革兰氏阴性菌,有研究表明其具有很强的产挥发性盐基氮和生物胺能力,因此其存在可能会加速食品的腐败,从而使发酵食品品质降低[27-28]。综上所述,本研究采集的部分兴山鲊广椒可能存在原料污染或发酵过程中密封环境遭到破坏等问题,从而使Pseudomonas快速生长繁殖。
本研究进一步在OTU水平上对兴山和保靖鲊广椒细菌类群进行了归类,并将所有样品中均存在的OTU称为核心OTU,其分析结果如图4所示。
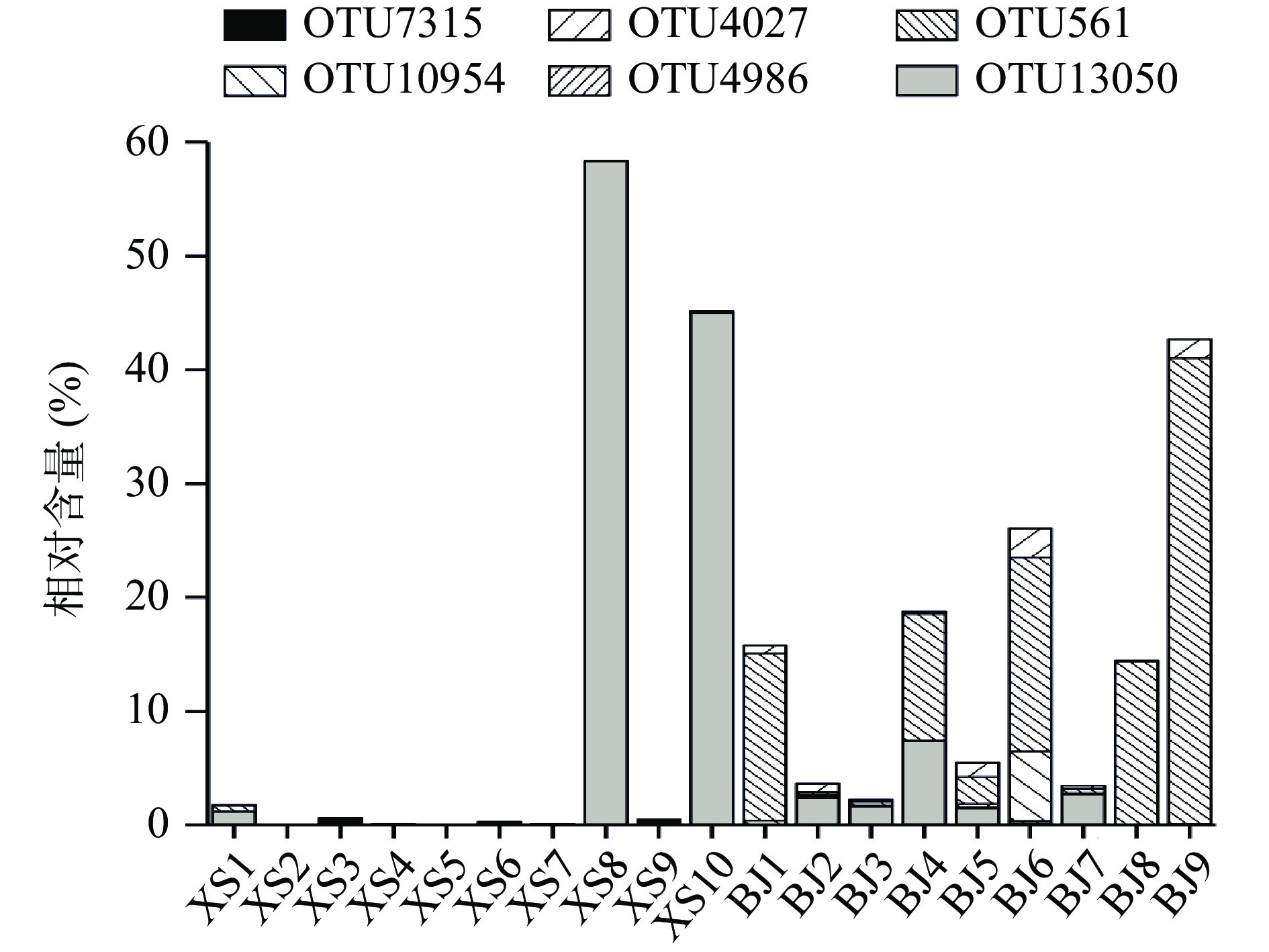
![]() 图 4 基于OTU水平的鲊广椒细菌类群结构分析Figure 4. Analysis of bacterial group structure of Zha-Chili based on OTU level
图 4 基于OTU水平的鲊广椒细菌类群结构分析Figure 4. Analysis of bacterial group structure of Zha-Chili based on OTU level由图4可知,兴山鲊广椒中的核心OTU相对较少,仅有一个隶属于Ralstonia(青枯菌属)的OTU7315,其平均相对含量为0.14%。保靖鲊广椒中共有5个核心OTU,分别为隶属于Pediococcus的OTU13050,平均相对含量为1.76%,隶属于Lactobacillus的OTU561、OTU4027、OTU10954和OTU4986,平均相对含量分别为11.26%、0.82%、0.81%和0.07%。超1指数显示兴山样本中细菌OTU数明显少于保靖,兴山与保靖地区鲊广椒细菌组成存在差异。
2.3 不同地区鲊广椒细菌群落结构的比较及LEfSe分析
本研究进一步采用统计学方法分析两地区鲊广椒菌群结构,其结果如图5所示。
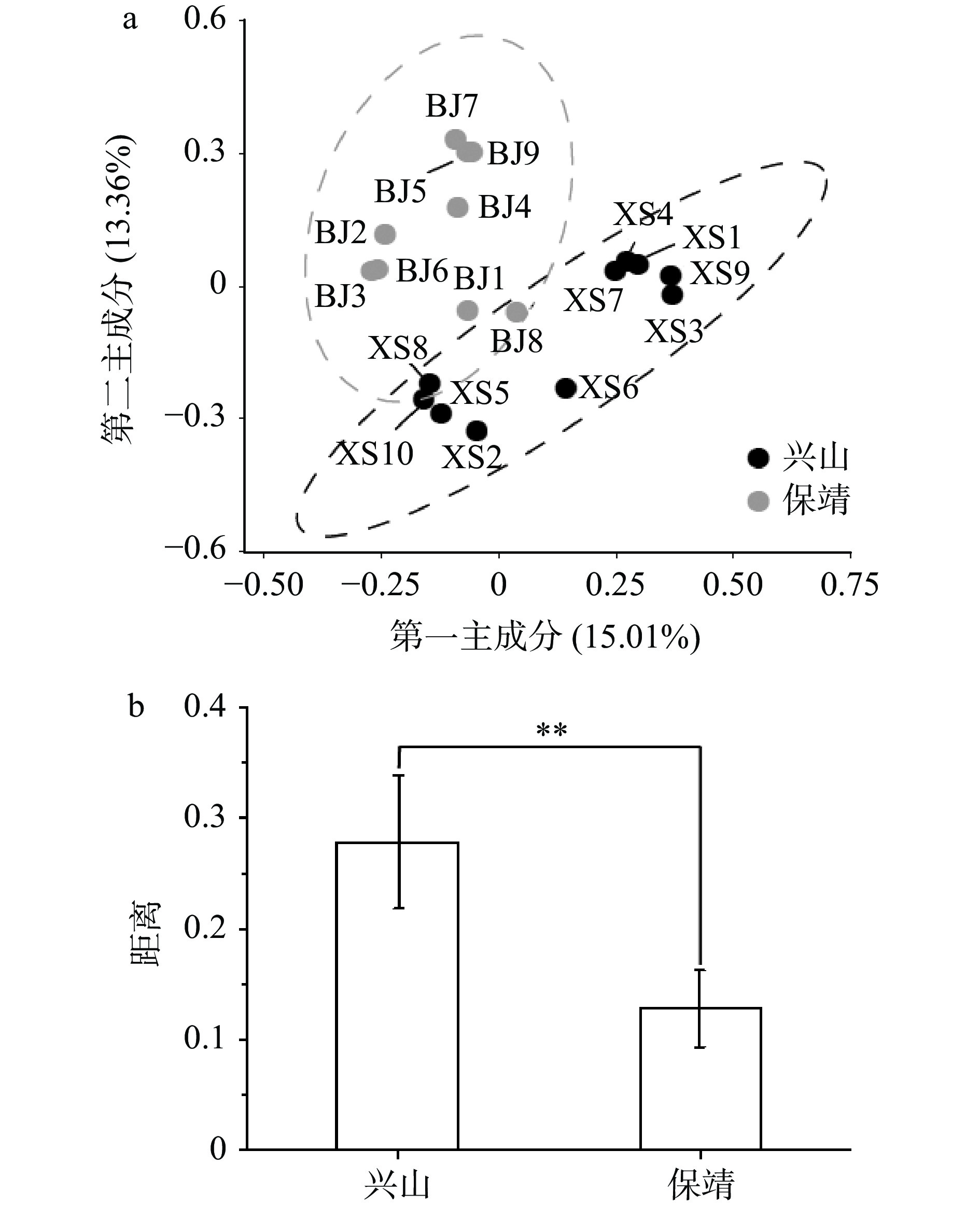
![]() 图 5 基于非加权的UniFrac 主坐标分析(a)和欧式距离分析(b)Figure 5. Unweighted UniFrac principal coordinate analysis (a) and Euclidean distance analysis (b)
图 5 基于非加权的UniFrac 主坐标分析(a)和欧式距离分析(b)Figure 5. Unweighted UniFrac principal coordinate analysis (a) and Euclidean distance analysis (b)由图5a可知,基于非加权的组间分析表明,不同地区鲊广椒样品在空间上呈现明显的分离趋势,而同地区鲊广椒样品则呈现明显的聚合趋势。通过统计学方法计算发现(图5b),兴山和保靖鲊广椒细菌群落结构在整体上存在显著差异(P<0.01)。
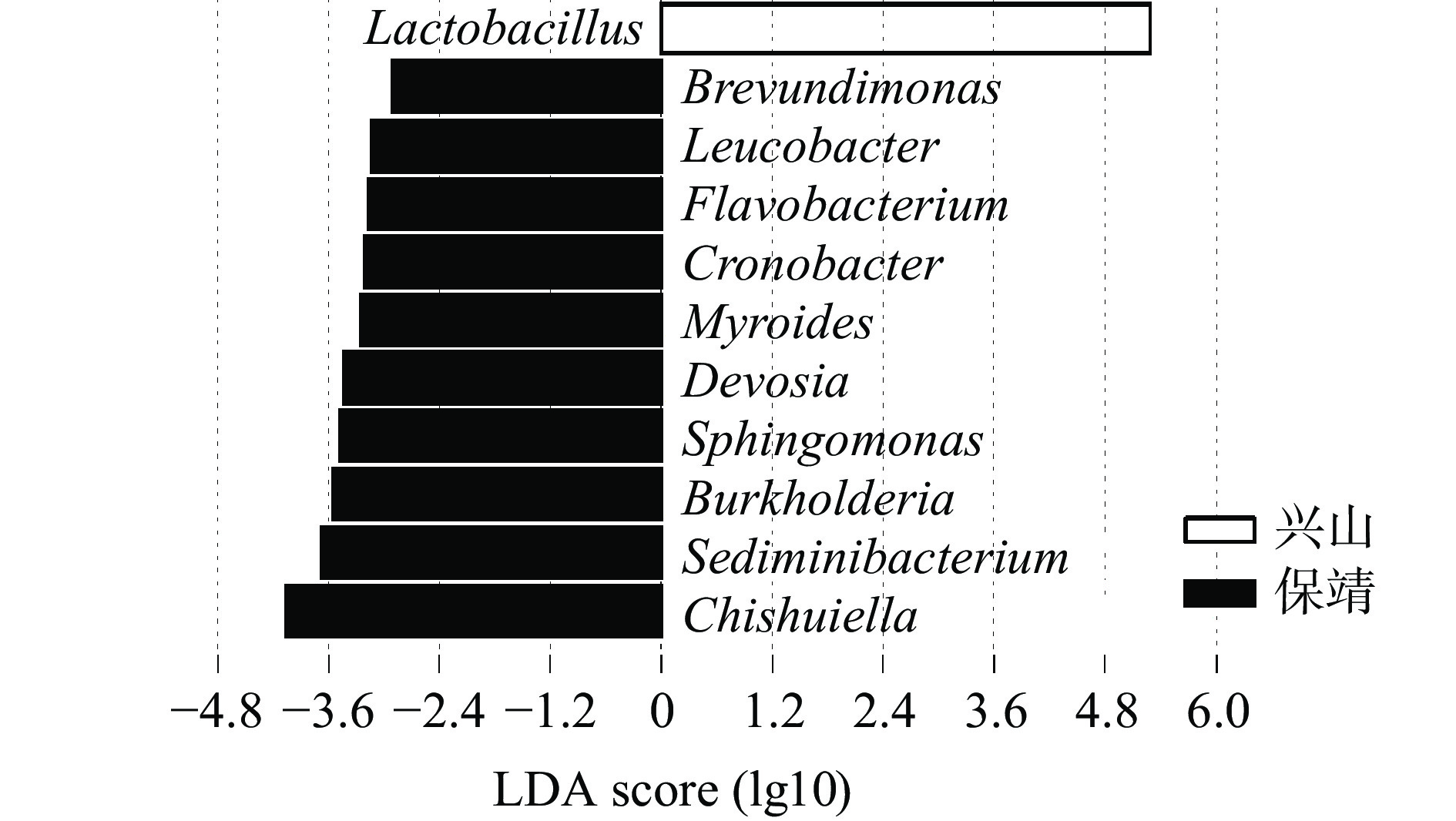
LDA分析是一种可以发现物种差异和标志物种的差异分析工具,其首先通过Kruskal-Wallis秩和检验对其丰度进行检测,找到组间差异显著的物种,然后使用Wilcoxon秩和检验对丰度差异显著的物种进行组间差异分析,最后使用线性判别分析对物种影响力进行评估[29]。为了进一步甄别兴山和保靖鲊广椒细菌群落结构的差异细菌类群,本研究使用LEfSe分析对样品进行统计,其结果如图6所示。
![]() 图 6 鲊广椒细菌类群LDA值分布柱状图Figure 6. Histogram of LDA value distribution of bacterial groups in Zha-Chili
图 6 鲊广椒细菌类群LDA值分布柱状图Figure 6. Histogram of LDA value distribution of bacterial groups in Zha-Chili由图6可知,当LDA score设为2时,有11个细菌属分布在图的两侧,分别为Lactobacillus、Brevundimonas(短波单胞菌属)、Leucobacter(白色杆菌属)、Flavobacterium(黄杆菌属属)、Cronobacter(阪崎肠杆菌属)、Myroides(香味菌属)、Devosia(德沃斯氏菌属)、Sphingomonas(单胞菌属)、Burkholderia(伯克氏菌属)、Sediminibacterium(沉降杆菌属)和Chishuiella(赤水河菌属),表明这些细菌属在两组样本间存在显著差异(P<0.01)。此外,兴山鲊广椒中Lactobacillus的LDA得分最高,而保靖鲊广椒中Chishuiella的得分最高,表明这两类细菌属对相应分组鲊广椒细菌组成影响最大。有研究表明,Chishuiella与腐乳产品中的酯类、酮类和醇类等物质的相关性较高,并提高发酵腐乳的香气[30]。
2.4 鲊广椒中微生物的表型预测分析
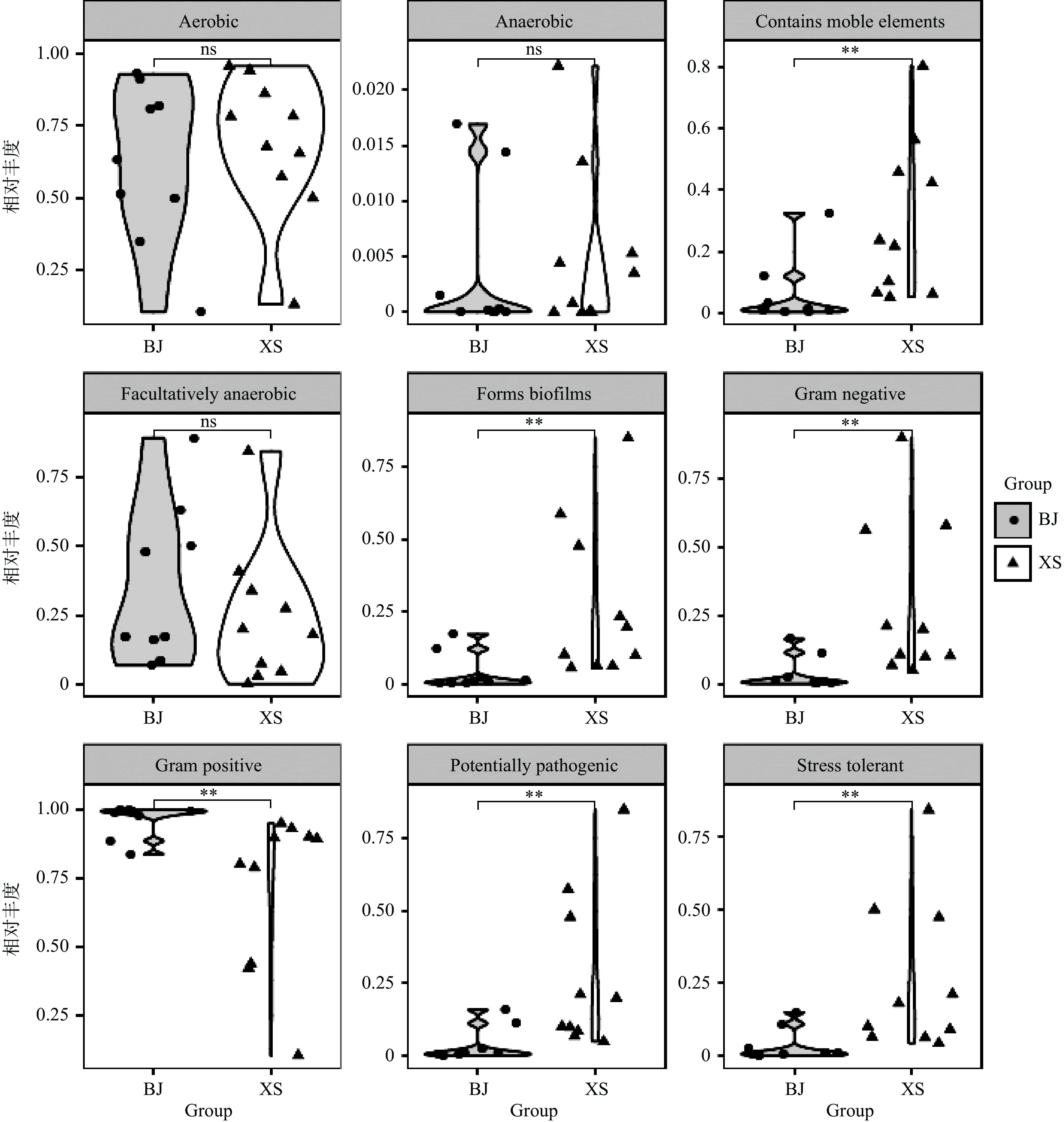
微生物群落及功能对鲊广椒品质有着重要影响,为探究菌群如何适应鲊广椒发酵过程中环境变化以及不同地区鲊广椒细菌特性,本研究进一步对兴山和保靖鲊广椒样品细菌类群的表型进行预测,其结果如图7所示。
由图7可知,兴山和保靖鲊广椒在移动原件含量、生物膜形成、革兰氏阴性、革兰氏阳性、致病潜力和氧化胁迫耐受等表型的表达具有显著差异(P<0.01),在其余表型上差异不显著(P>0.05)。由此可知,兴山鲊广椒的细菌类群在移动原件含量、生物膜形成、革兰氏阴性、致病潜力和氧化胁迫耐受等表型的表达显著高于保靖鲊广椒,而在革兰氏阳性的表达上则呈现相反趋势,说明兴山鲊广椒细菌类群更容易适应环境的变化。
3. 结论
本研究分析兴山和保靖共19份鲊广椒样品,经研究发现,兴山鲊广椒细菌菌群丰度和多样性均显著低于保靖鲊广椒(P<0.05)。两地区鲊广椒样品均有为Lactobacillus、Pediococcus和Weissella等优势细菌属,其中兴山鲊广椒的优势细菌还有Pseudomonas,且经检验发现,兴山鲊广椒中Lactobacillus的含量显著低于保靖鲊广椒,Pseudomonas则呈现相反的趋势。兴山和保靖鲊广椒样品间具有一定的空间距离(P<0.01),同时发现11个具有显著差异的细菌类群,其中得分最高的为Lactobacillus和Chishuiella。表型预测发现,兴山鲊广椒的细菌类群较保靖鲊广椒来说可能更能适应环境的变化。
-
![]()
图 1 香农和超1指数的箱形图
注:*表示差异显著(P<0.05),**表示差异非常显著(P<0.01),***表示差异极显著(P<0.001);图5同。
Figure 1. Shannon and Chao 1 index box graph
![]()
图 2 基于门水平的鲊广椒细菌多样性分析
Figure 2. Analysis of bacterial diversity of Zha-Chili based on phylum level
![]()
图 3 基于属水平的鲊广椒细菌多样性分析
Figure 3. Bacterial diversity analysis of Zha-Chili based on genus level
![]()
图 4 基于OTU水平的鲊广椒细菌类群结构分析
Figure 4. Analysis of bacterial group structure of Zha-Chili based on OTU level
![]()
图 5 基于非加权的UniFrac 主坐标分析(a)和欧式距离分析(b)
Figure 5. Unweighted UniFrac principal coordinate analysis (a) and Euclidean distance analysis (b)
![]()
图 6 鲊广椒细菌类群LDA值分布柱状图
Figure 6. Histogram of LDA value distribution of bacterial groups in Zha-Chili
-
[1] YÉPEZ A, LUZ C, MECA G, et al. Biopreservation potential of lactic acid bacteria from Andean fermented food of vegetal origin[J]. Food Control,2017,78(3):393−400.
[2] 王玉荣, 沈馨, 董蕴, 等. 鲊广椒细菌多样性评价及其对风味的影响[J]. 食品与机械,2018,34(4):25−30. [WANG Y, SHEN X, DONG Y, et al. Characterization of bacterial microflora and their functions on flavor quality[J]. Food & Machinery,2018,34(4):25−30. doi: 10.13652/j.issn.1003-5788.2018.04.005 [3] 崔小利, 王薇, 阚建全. 鲊辣椒纯种发酵的菌株优选[J]. 食品科学,2014,35(21):149−153. [CUI X, WANG W, KAN J. Screening strains for use in pure culture fermentation for the production of Zha-Chili, a traditional chinese fermented hot pepper product[J]. Food Science,2014,35(21):149−153. doi: 10.7506/spkx1002-6630-201421029 [4] 李娜, 张苗苗, 舒娜, 等. 咸丰和当阳地区鲊广椒细菌群落结构差异性研究[J]. 中国酿造,2020,39(10):48−53. [LI N, ZHANG M, SHU N, et al. Difference of bacterial community structure of zha-chili in xianfeng and dangyang region[J]. China Brewing,2020,39(10):48−53. doi: 10.11882/j.issn.0254-5071.2020.10.010 [5] 韦明明. 番茄酸汤发酵过程分析及混菌发酵工艺研究[D]. 南京: 南京农业大学, 2016. WEI M. Analysis of tomato sour soup during fermentation process and study on the mixed culture fermentation technology for tomato sour soup[D]. Nanjing: Nanjing Agricultural University, 2016.
[6] 荆雪娇. 传统发酵蔬菜微生物群落结构分析[D]. 太原: 山西大学, 2016. JING X. Microbial community structure analysis of traditional fermented vegetables[D]. Taiyuan: Shanxi University, 2016.
[7] SUN X, LYU G, LUAN Y, et al. Analyses of microbial community of naturally homemade soybean pastes in Liaoning Province of China by Illumina MiSeq Sequencing[J]. Food Research International,2018,111(9):50−57.
[8] XIE M, AN F, WU J, et al. Meta-omics reveal microbial assortments and key enzymes in bean sauce mash, a traditional fermented soybean product[J]. Journal of the Science of Food and Agriculture,2019,99(14):6522−6534. doi: 10.1002/jsfa.9932
[9] LEMBELLA B A E, LEBONGUY A A, GOMATCHIMBAKALA J, et al. Bacterial community diversity of fermented pepper in brazzaville revealed by Illumina MiSeq of 16S rRNA gene[J]. Food And Nutrition Sciences,2021,12(1):37−53. doi: 10.4236/fns.2021.121004
[10] DWA B, GONG C, YAO T, et al. Effects of temperature on paocai bacterial succession revealed by culture-dependent and culture-independent methods- ScienceDirect[J]. International Journal of Food Microbiology,2020,317(4):1−31.
[11] AGNIESZKA, PIOTROWSKA-CYPLIK, KAMILA, et al. Characterization of specific spoilage organisms (SSOs) in vacuum-packed ham by culture-plating techniques and MiSeq next-generation sequencing technologies[J]. Journal of the Science of Food and Agriculture,2016,97(2):659−668.
[12] 郭壮, 葛东颖, 尚雪娇, 等. 退化和正常窖泥微生物多样性的比较分析[J]. 食品工业科技,2018,39(22):93−98. [GUO Z, GE D, SHANG X, et al. Comparative analysis on the diversity of bacterial microflora in degenerated and normal pit mud[J]. Science and Technology of Food Industry,2018,39(22):93−98. doi: 10.13386/j.issn1002-0306.2018.22.018 [13] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods,2010,7(4):335−336.
[14] KNIGHT R. PyNAST: A flexible tool for aligning sequences to a template alignment[J]. Bioinformatics,2010,26(2):266−267. doi: 10.1093/bioinformatics/btp636
[15] GOLOB J L, MARGOLIS E, HOFFMAN N G, et al. Evaluating the accuracy of amplicon-based microbiome computational pipelines on simulated human gut microbial communities[J]. Bmc Bioinformatics,2017,18(1):283. doi: 10.1186/s12859-017-1690-0
[16] KNIGHT R. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics,2011,27(16):2194−2200. doi: 10.1093/bioinformatics/btr381
[17] DESANTIS T Z, HUGENHOLTZ P, LARSEN N, et al. Greengenes, a chimera-checked 16s rRNA gene database and workbench compatible with ARB[J]. Applied & Environmental Microbiology,2006,72(7):5069−5072.
[18] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools[J]. Nucleic Acids Research,2012,41(D1):590−596. doi: 10.1093/nar/gks1219
[19] COLE J R, WANG Q, CARDENAS E, et al. The ribosomal database project: Improved alignments and new tools for rRNA analysis[J]. Nucleic Acids Research,2009,37(suppl_1):141−145.
[20] LOZUPONE C, KNIGHT R. UniFrac: A new phylogenetic method for comparing microbial communities[J]. Applied and Environmental Microbiology,2005,71(12):8228−8235. doi: 10.1128/AEM.71.12.8228-8235.2005
[21] WARD T, LARSON J, MEULEMANS J, et al. BugBase predicts organism level microbiome phenotypes[J]. BioRxiv, 2017.
[22] WANG H L, HOPFER H, COCKBURN D W, et al. Characterization of microbial dynamics and volatile metabolome changes during fermentation of Chambourcin hybrid grapes from two Pennsylvania regions[J]. Frontiers in Microbiology,2021,11(1):3454−3475.
[23] CHEN H, ZHANG Q, WAN H, et al. Effect of total inoculum size containing Lactobacillus acidophilus or Lactobacillus casei on fermentation of goat milk[J]. Advance Journal of Food Science and Technology,2015,7(3):183−186. doi: 10.19026/ajfst.7.1290
[24] 赵跃, 李春生, 王悦齐, 等. 罗非鱼鱼糜自然发酵过程中微生物群落结构对其品质形成的影响[J]. 食品科学,2021,42(18):119−126. [ZHAO Y, LI C, WANG Y, et al. Effect of microbial community structure on quality formation of naturally fermented tilapia surimi[J]. Food Science,2021,42(18):119−126. doi: 10.7506/spkx1002-6630-20200915-194 [25] BORGONOVI T F, CASAROTTI S N, PENNA A L B. Lacticaseibacillus casei SJRP38 and buriti pulp increased bioactive compounds and probiotic potential of fermented milk[J]. LWT,2021,143:111124. doi: 10.1016/j.lwt.2021.111124
[26] BAS T, DOUWE M. Systems biology of lactic acid bacteria: For food and thought[J]. Current Opinion in Systems Biology,2017,6(12):7−13.
[27] 王航. 草鱼贮藏过程中品质变化规律及特定腐败菌的研究[D]. 北京: 中国农业大学, 2016. WANG H. Quality changes and the specific spoilage organisms of grass carp (Ctenopharyngodon idellus) fillets during storage[D]. Beijing: China Agricultural University, 2016.
[28] RAWAT S. Food spoilage: Microorganisms and their prevention[J]. Asian Journal of Plant Science and Research,2015,5(4):47−56.
[29] SEGATA N, IZARD J, WALDRON L, et al. Metagenomic biomarker discovery and explanation[J]. Genome Biology,2011,12(6):R60. doi: 10.1186/gb-2011-12-6-r60
[30] 孙娜, 张雅婷, 于寒松, 等. 发酵型青腐乳菌群结构与风味物质及其相关性分析[J]. 食品科学,2020,41(22):177−183. [SUN N, ZHANG Y, YU H, et al. Microflora structure and flavor components and correlation between them in fermented stinky tofu[J]. Food Science,2020,41(22):177−183. doi: 10.7506/spkx1002-6630-20190929-360 -
期刊类型引用(6)
1. 许津阁,郑卓琦,侯鹏颉,马高兴,熊彦娣,马壮,刘萌,赵靓,廖小军. 不同产地酱用卡宴辣椒原料品质评价. 食品工业科技. 2025(01): 317-332 .  本站查看
本站查看
2. 熊岑,阮沛仪,郭晓刚,刘大千,曾丽娴. 辣椒中多酚的提取工艺和抗氧化活性研究. 中国调味品. 2024(02): 89-94 . 百度学术
3. 金怀慷,杨灿,刘力. 云南5种辣椒加工适应性分析与比较. 天津农业科学. 2024(06): 77-82 . 百度学术
4. 田筱,涂德辉,梁传静,王永平,李伟,邢丹. 贵州五地辣椒矿物质含量及果实品质分析. 北方园艺. 2024(18): 7-15 . 百度学术
5. 杨娅,吴康云,黄冬福,周鹏,付文婷,王楠艺,何建文. 基于主成分分析对不同地区辣椒品质的综合评价. 食品工业科技. 2024(23): 264-271 . 本站查看
6. 付文婷,王楠艺,周鹏,杨娅,彭世清,黄冬福,何建文. 不同产区辣椒果实品质差异及其与气象因子的相关性. 南方农业学报. 2024(09): 2763-2771 . 百度学术
其他类型引用(1)
 下载:
下载:

 下载:
下载:






计量
- 文章访问数: 157
- HTML全文浏览量: 32
- PDF下载量: 7
- 被引次数: 7



