


Advances in Methods of Microbial Molecular Ecology
-
摘要: 微生物分子生态学是分子生物学与微生物生态学交叉而形成的学科,对于认识微生物生态系统组成结构、功能、微生物之间以及微生物与环境之间的相互关系具有重要意义。本文以微生物分子生态学研究方法作为切入点,对目前广泛使用的微生物分子生态学研究技术和方法进行综合介绍、分析和总结,对各种方法的优缺点和应用进行评论,以期为认识和调控生态系统的微生物群落提供参考。Abstract: Microbial molecular ecology is an interdisciplinary of molecular biology and microbial ecology. It is important to understand the composition and structure, function, the relationship between microorganisms and the relationship between microorganisms and the environment in microbial ecosystems. Microbial molecular ecology research techniques and methods widely used are comprehensively introduced, analyzed and summarized, and the advantages, disadvantages and applications of the techniques and methods are discussed. This study would provide a reference for understanding and regulating the kind and population of microbial communities in ecosystems.
-
微生物生态学是基于微生物群体的科学,利用微生物群体DNA/RNA等标志物,重点研究微生物群落构建、组成演变、多样性及其与环境的关系,在生态学理论的指导和反复模型拟合下由统计分析得出具有普遍意义的结论[1]。随着研究的深入,传统的技术在非可培养微生物研究方面存在一定局限性,分子生物学研究技术被借鉴到微生物生态学研究中,在物种遗传多样性、分子适应性、变异分子机制及其进化意义等基础理论方面取得了突破[2]。
目前环境微生物生态学主要研究环境中存在哪些微生物和分析微生物在环境中的作用两个方面[3],此外,微生物之间以及微生物与环境因子之间的相互作用受到越来越多的关注[4]。近十几年来,微生物多样性以及群落结构的研究一直沿用传统分离、培养、鉴定并描述特征的方法,然而,研究证实自然界中有85%~99.9%的微生物至今还不可纯培养[5]。随着环境微生物DNA提取方法的不断改进,以核酸为依据对微生物进行分类方面的研究获得了较大发展,涌现出许多微生物分子生态学研究方法,如变性梯度凝胶电泳、末端限制性长度多态性、荧光原位杂交技术、实时荧光定量PCR技术、扩增子测序技术、宏基因组学、宏转录组学、功能基因组学、基因芯片等[6],常用微生物分子生态学研究方法的优缺点见表1。
表 1 微生物分子生态学研究方法比较Table 1. Comparison of research methods of microbial molecular ecology类别 代表技术 优点 缺点 应用
案例
传
统
方
法分离培养法 简便易行 只能分离有限种类的微生物 [7] Biolog微平板法 可重复且容易使用,可以产生大量
反映群落代谢特征的数据只能检测到一些代谢活性高和可培养的细菌,不能反映真菌和生长缓慢的细菌,检测结果会受到接种密度的干扰 [8] PLFA图谱分析 简单快速,不需要特殊的设备,
对菌群环境变化敏感[9]分类分辨率差,结果受提取方法和菌群pH动态变化的影响,会产生一定的误差 [10]
D
N
A
指
纹
图
谱变性梯度凝胶电泳(DGGE) 简单,无放射性,相对无毒,检测率
和特异性高,杂合子检测率高分析高GC含量和高长度片段困难,分析费时费力且费用高,PCR引物不同会引起一定的误差 [11] 末端限制性长度多态性(T-RFLP) 可产生大量重复且分辨率高的结果,数据具体化,易实现自动化检测,操作简便,灵敏度高 无法区分近缘种微生物,不能确定各类微生物的绝对含量,片段长度范围的确定、噪声峰的去除及大小相近的片段合并都会对结果造成影响 [12]
荧光
标记荧光原位杂交技术(FISH) 经济、安全且探针稳定,实验周期短、特异性好、定位准确,可定位1 kb长度的DNA等 杂交效率低且检测灵敏度不高 [13] 实时荧光定量PCR 快速精确,解决了不可培养和未知微生物的定量问题,可以达到种的水平 无法得到整个微生物的群落的信息,实验难度大,
对实验结果有影响[14]
高
通
量
测
序
扩增子测序测序片段短,无需构建基因文库,高输出量和高解析度且测序结果覆盖整个微生物群落的信息 仪器设备昂贵,测序成本高,只能获得样品中微生物的相对丰度,无法确定样品中微生物的具体数量,对于16S rRNA测序来说,需要对样本进行PCR扩增,将会使结果
产生一定的误差[15]
宏基因组学高分辨率,没有因使用引物和PCR而产生的误差,直接对全部DNA进行测序,节省了
许多不必要的步骤[16]
需要一定的生物信息学知识储备和分析设备[17]
宏转录组学可以检测完整的转录组(编码和非编码RNA),可以检测RNA序列的变体和异构体,不需要微生物
基因组的知识储备样品制备步骤比较繁杂且样本的数量较少,需要经过
复杂的数据分析才可得到最终的实验结果[18] 功能
基因组学基因芯片 灵敏度高,可直接对高通量序列信息进行分析 低同源序列的交叉杂交导致分析结果产生偏差 [19] RNA测序 可以在单细胞水平上对细胞群进行特异性分析 对低丰度转录本的检测性能有待提高 [20] 本文基于前人的研究工作,综述了微生物分子生态学的研究方法,以期为客观地认识、分析和利用微生物群落乃至微生物生态系统提供技术参考。
1. 传统方法
常用微生物分子生态学研究方法中的分离培养法、Biolog微平板法和PLFA图谱分析均属于传统方法,即环境微生物数据是通过分析微生物生长的物质获得的,如液体培养物中的细胞或通过涂布获得的菌落,以及微生物代谢所产生的物质和微生物自身的组成部分。
1.1 分离培养法
传统微生物培养法[21]是将所获得样本中的微生物进行分离、提纯、鉴定后通过肉眼观察其特征,以此来区分样本中菌群的结构。由于传统的纯培养法只能在实验室进行,且仅能区分环境中低于10%的微生物种类,因而得到的结果不能完全反映环境微生物具体的生态多样性,且该方法耗时较长。
1.2 Biolog微平板法
Biolog微平板法是通过微生物利用碳源进行呼吸产生NADH,并与底物中的还原染料发生反应所产生的显色效应来鉴别菌种。Biolog微平板法可用于环境微生物群落的研究,Ei-Liethy等[22]分别使用Biolog Gen III系统和聚合酶链式反应分离和鉴定水槽排水管中的芽孢杆菌,证实PCR对枯草芽孢杆菌的鉴定比Biolog更敏感,采用分子方法可以弥补Biolog微平板法在鉴定细菌分离物上的不足。Morgan等[23]使用159株临床分离株对Biolog鉴定系统和16S核糖体RNA基因测序方法进行了评估,结果表明:16S rRNA基因测序提供了更准确的非典型细菌鉴定方法,对于鉴定挑剔的革兰氏阴性杆菌类别的生物时,确定Biolog系统存在缺陷。Biolog微平板法具有以下特点[24]:测定简便,具有可重复性;无需分离培养纯种环境微生物;Biolog平板中的碳源和介质的pH可能不能代表土壤中存在的碳源和pH情况;不能全面了解样本中微生物群落结构,还应采用其他研究方法。
1.3 PLFA图谱分析
PLFA图谱分析又称磷脂脂肪酸图谱分析,它将磷脂作为存活微生物群落的标记物,利用气相色谱分析,并与已知数据库(厌氧菌库、酵母菌库等)对比即可得到环境样品中微生物的生物量和群落结构信息。
PLFA图谱分析可用于土壤种群多样性的研究。Chen等[25]在研究模拟的春季降水后土壤微生物群落组成的短期变化过程中发现基于不同微生物生物标记物(PLFA与DNA)方法的组合使用,结果却相互一致,但基于PLFA的方法更为敏感,基于DNA的方法则可以提供更精细的微生物类群信息。Orwin等[26]测试了PLFA剖面法和16S rRNA基因元编码这两种技术在量化五种土地利用之间细菌群落结构差异的相对能力,结果发现虽然两种方法得出的结果具有一致性,但是16S rRNA基因元编码在灵敏度上将更胜一筹。该方法突破了原有的微生物分离培养的限制,依靠简单快速的分析程序和不使用特殊的设备,赢得了广大研究者们的青睐,但是仅限于属的水平,且受到提取方法和环境pH动态变化的影响,所以用于特异微生物类群的鉴定结果还不够准确[27],应结合其他方法以提高鉴定结果的准确性。
2. DNA指纹图谱法
DNA指纹图谱法即使用基因探针与DNA酶切片段进行杂交,获得由多个位点上的等位基因组成的长度不等的杂交带图纹,并通过数据分析得出结论。
2.1 变性梯度凝胶电泳(DGGE)
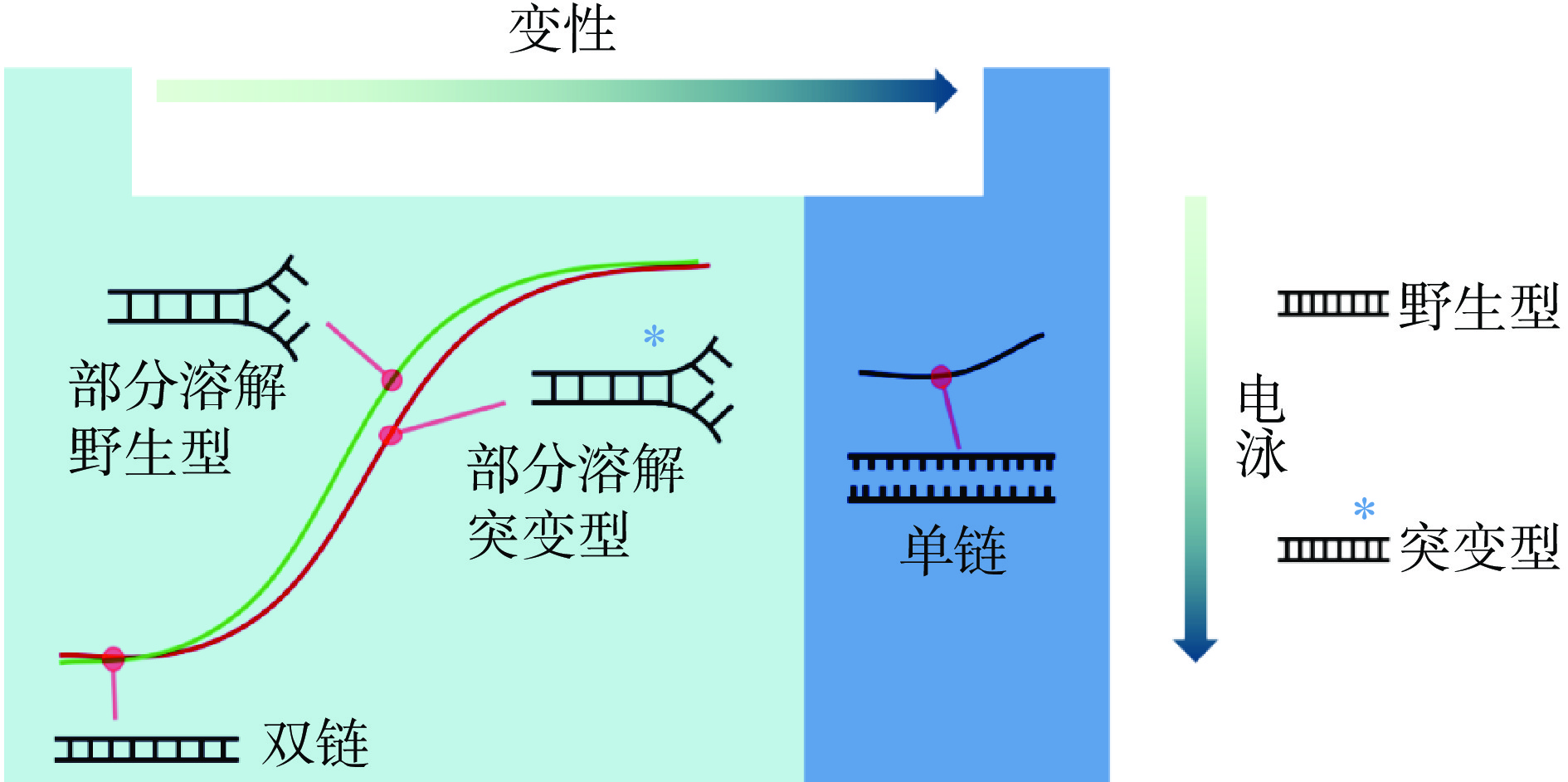
变性梯度凝胶电泳(DGGE)因具有高效且操作简单的特点,极大地拓展了对环境中不可培养微生物的发现,但是它具有谱带重叠的缺陷,导致其成为一种低分辨率的分析方法[28],因此该方法不是一种普适的分析方法,只能作为研究菌群结构的辅助手段之一[29],故需要结合指纹图谱技术和测序技术作进一步的鉴定分析,后者通过克隆、测序建立微生物区系的16S rRNA/rDNA文库。通过系统发育分析,建立进化树,从而获得微生物多样性分析,但这种方法相对于指纹图谱技术来说,价格昂贵。其原理见图1[30],该方法根据DNA的解链特性,不同碱基组成的DNA在聚丙烯酰胺凝胶电泳时会产生不同的电泳迁移速率,从而使不同DNA分子得以分离。
![]() 图 1 变性梯度凝胶电泳(DGGE)原理图Figure 1. Schematic diagram of denaturing gradient gel electrophoresis (DGGE)
图 1 变性梯度凝胶电泳(DGGE)原理图Figure 1. Schematic diagram of denaturing gradient gel electrophoresis (DGGE)PCR-DGGE技术因在提取DNA后能够直接提供环境样本中细菌群落的指纹而被微生物生态学广泛应用,Xiong等[31]使用DGGE和高通量测序(HTS)来研究婴儿配方粉生产主要阶段中的细菌群落结构和分布,虽然HTS和DGGE在反映优势菌群变化方面上不分伯仲,但是HTS在反映细菌群落丰富度方面上更胜一筹。Chahorm等[32]验证了PCR-DGGE在快速检测食品中弧菌种类上的可行性。Bo等[33]利用扩增子测序技术和DGGE对缅甸传统发酵茶叶中细菌和真菌的多样性进行分析,虽然两种方法在该项研究中具有很高的相似性,但是DGGE因很难检测到小于104 CFU/g样品中微生物的种群,导致其多样性分析结果较差。PCR-DGGE技术是微生物分子生态学技术中最常用的方法之一,但该方法与高通量测序技术等其他方法相比,在检测丰度和检测限度上具有明显的缺点,同时样本的处理、DNA的提取、PCR扩增的缺陷都会影响检测结果,故需要结合其他分子生物手段进行分析。
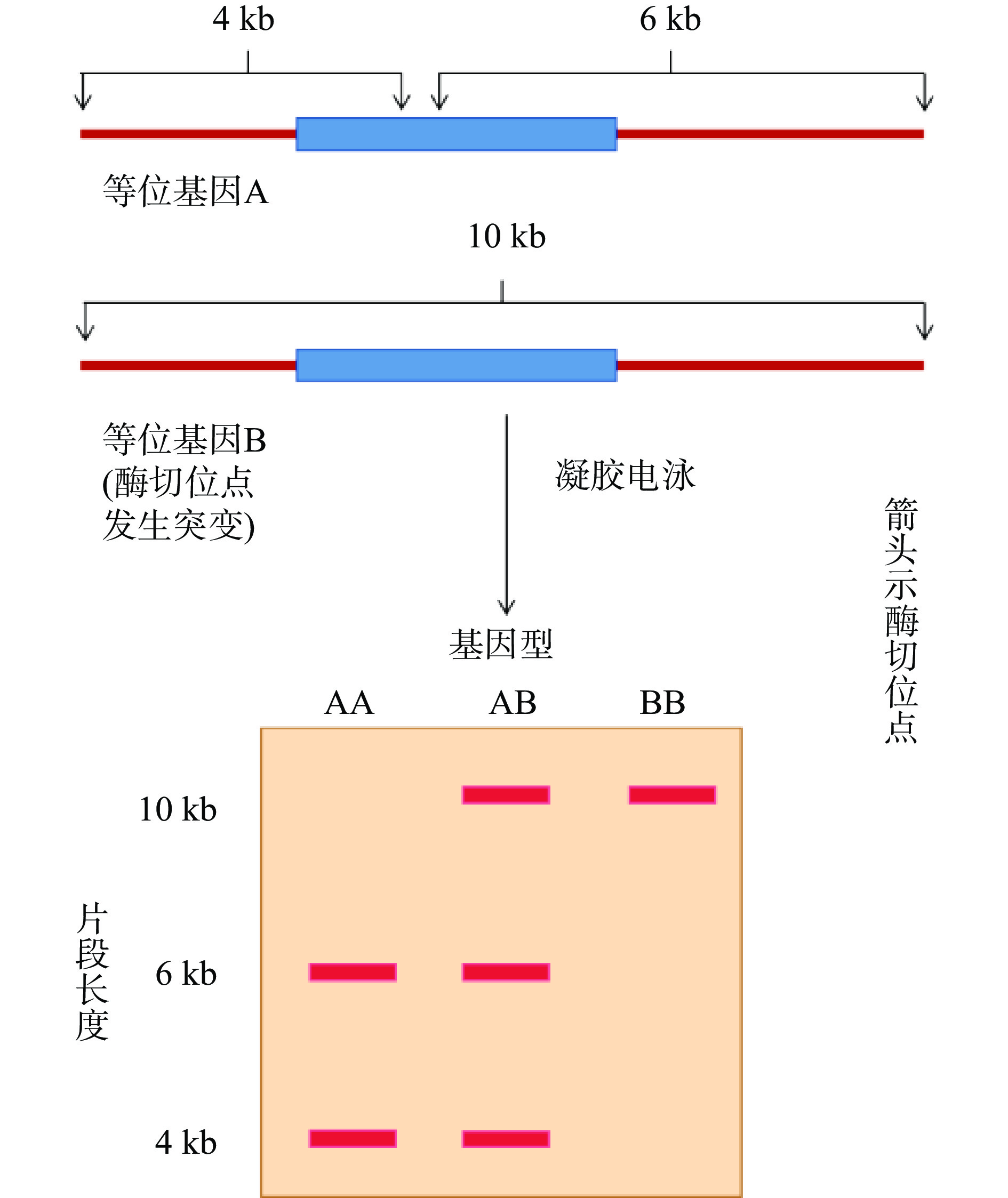
2.2 末端限制性长度多态性(T-RFLP)
末端限制性长度多态性又称为16S rRNA基因的末端限制性片段分析技术,其原理见图2[34],基于限制性内切酶的特异性——只在识别位点切割双链DNA,因此具有高度的序列特异性和较高的分辨率、精确度和灵敏度等特点,且能够迅速获得大量数据,准确、快速、真实地反映微生物的多样性。
![]() 图 2 末端限制性长度多态性原理图Figure 2. Schematic diagram of terminal restricted length polymorphism
图 2 末端限制性长度多态性原理图Figure 2. Schematic diagram of terminal restricted length polymorphismT-RFLP技术近年来被广泛应用于环境微生物群落多样性的分析中。López等[35]在区分蜂蜜和其他蜂源中80个菌株的过程中,证实RFLP方法简单易行,能用于蜂蜜样品中菌种的预筛选和分离鉴定。Rosa等[36]分别使用API 20 Strep(链球菌鉴定系统)、PCR-RFLP和MALDI-TOF-MS(基质辅助激光解吸电离飞行时间质谱)分析了从绵羊和山羊奶样品中收集的195种链球菌分离株,结果显示PCR-RFLP是鉴定牛奶样品中链球菌最准确的方法。虽然T-RFLP具有存在不同菌种DNA的差异性扩增、菌种间目标DNA拷贝数存在差异、复杂群落多样性被低估等缺点,但在使用T-RFLP法进行分析时,充分考虑研究目的、环境条件和样品特征,便会使分析结果准确可靠[37],因此T-RFLP技术在操作性和准确性方面与其他技术相比具有一定的优势。
3. 荧光标记法
荧光标记法[38]是利用荧光蛋白或荧光蛋白基因作为标志物对研究对象进行标记的分析方法,该方法所依赖的化合物称为荧光物质,利用荧光检测装置定位荧光所在位置或分析荧光强弱来达到实验目的。
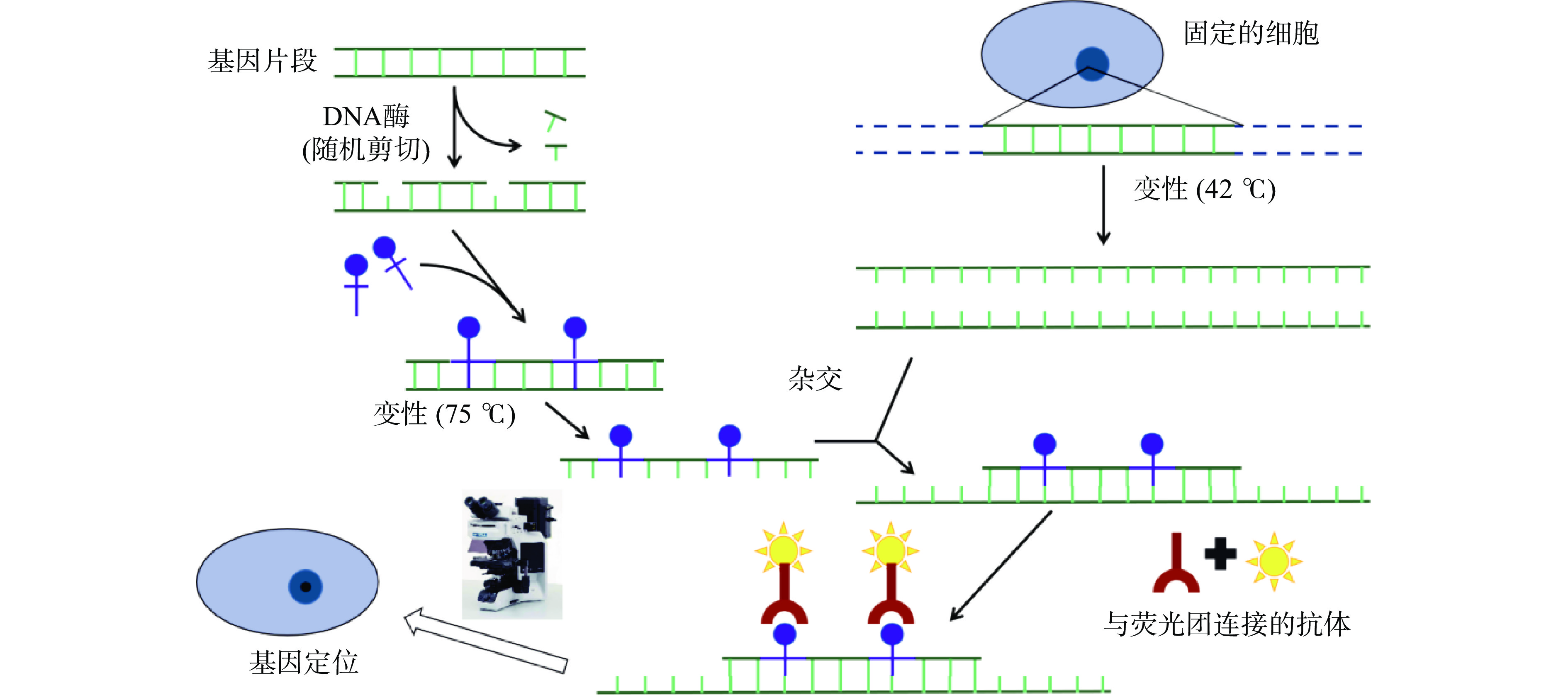
3.1 荧光原位杂交技术(FISH)
荧光原位杂交技术(FISH)[39]是一种在细胞环境中对核酸进行空间检测和定量的技术。原理见图3[40],用荧光素标记的核酸探针与待测样本的核酸序列按碱基互补配对原则进行杂交,最终可以用荧光显微镜观察到基因所在位置。作为一种非放射性的检测系统,因其具有经济、安全且探针稳定,实验周期短、特异性好、定位准确且可定位1 kb长度的DNA等优点[41],现已成为用于细胞和组织上转录分析的强大细胞遗传学分析方法,但由于细胞内靶定分子数量较少,且探针在细胞内的渗透性差,杂交效率低等因素,导致检测灵敏度不高,同时可能会产生假阴性或假阳性,适当的使用阴性和阳性对照以及开发可靠的测定方案是解决问题的关键[42]。
荧光原位杂交技术可应用于医学、环境学、微生物学等各大领域。Zhang等[43]利用基于分子信标的荧光杂交(MB-FISH)用于快速直接检测金黄色葡萄球菌,结果显示MB-FISH具有很强的特异性和高灵敏度,可用于在阳性血液培养物中直接检测金黄色葡萄球菌。Salimi等[44]在温度、探针、盐和甲酰胺浓度等参数上优化FISH效率的过程中,证实FISH是一种快速且准确地检测切碎的羔羊沙门氏菌的可靠方法。Baliga等[45]发现应用于培养的MN Genus-MTBC双探针荧光FISH分析也可用于资源有限的结核病流行国家,以快速鉴定和区分痰液样本中的MTBC(结核分枝杆菌复合体)和NTM(非结核分枝杆菌)。荧光原位杂交虽然在技术上存在一定的缺陷,但可以通过优化温度、探针、荧光素颜色以及DNA浓缩程度等条件,提高荧光原位杂交的分辨率,使其可以快速准确地检测待测样本。
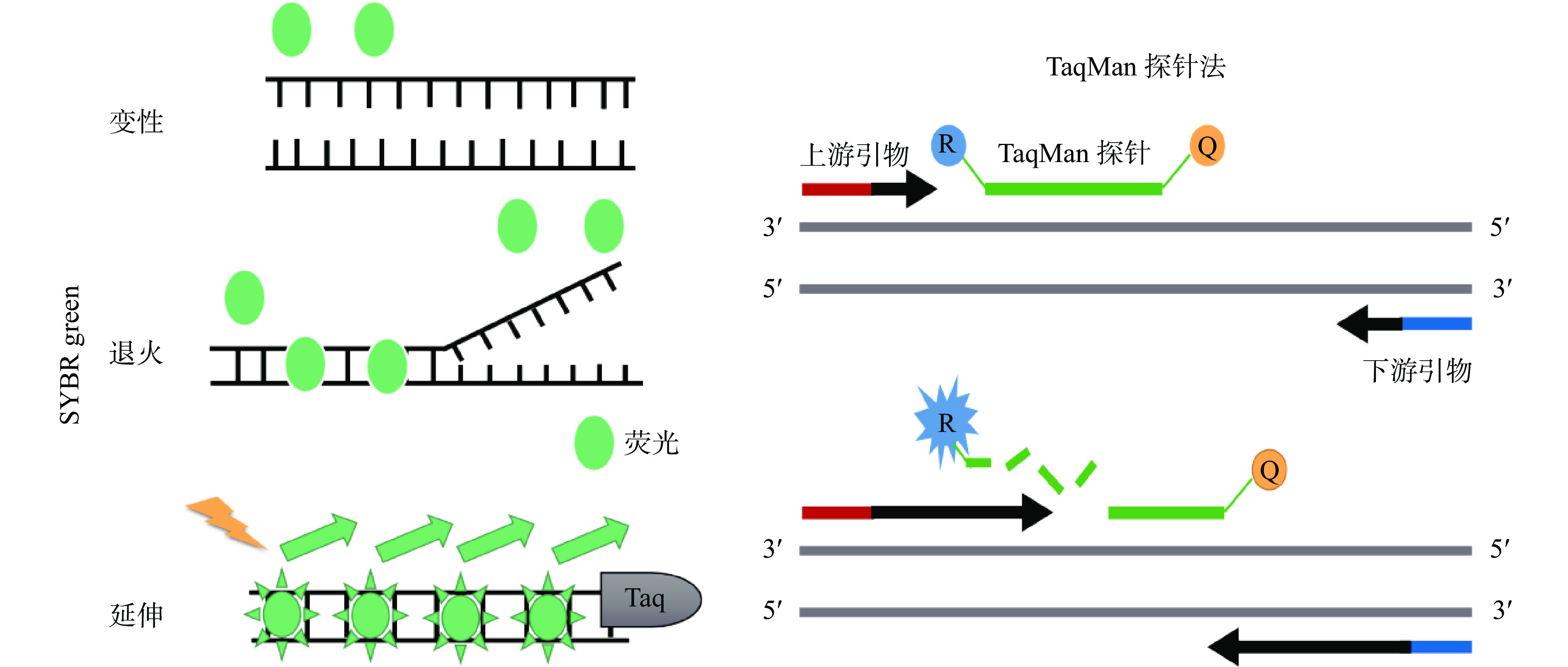
3.2 实时荧光定量PCR技术(RT-qPCR)
实时荧光定量PCR技术分为染料法和探针法,其原理见图4[46],在PCR反应中加入荧光基团,通过连续监测荧光信号出现的先后顺序以及信号强弱的变化,即时反映出目的基因的初始量。该方法适用于任何DNA,并且不必设计复杂的探针,同时具有非常灵敏且便宜的优点,虽极易与非特异性双链DNA结合,产生假阳性,但具体研究中可以通过熔解曲线的分析,优化反应条件来解决假阳性问题。
![]() 图 4 实时荧光定量PCR原理图Figure 4. Principle diagram of real-time fluorescence quantitative PCR
图 4 实时荧光定量PCR原理图Figure 4. Principle diagram of real-time fluorescence quantitative PCR实时荧光定量PCR分为绝对定量和相对定量,分别可应用于病原体检测、转基因食品检测、基因表达研究、高通量基因检测方法的验证、药物疗效考核和耐药性研究等方面。Bahlinger等[47]建立了两种多重qPCR方法来检测肉类和热处理肉制品中假单胞菌、肠杆菌科、热链丝菌和葡萄球菌的含量,有助于筛查涉嫌食品欺诈的食品。Pancza等[48]使用qPCR法对牛奶中的致病菌和腐败菌进行了检测,验证了该方法的可行性。Brotons等[49]利用RT-qPCR检测巴塞罗那地区2709名无症状青少年和成年人的唾液样本,仅有0.6%的检测结果无效,证实RT-qPCR是一种可以简单、快速且准确地检测无症状患者是否感染新型冠状病毒,并有望在社区扩大筛查中推广应用。RT-qPCR因其具有快速且准确的特点,受到广大研究者的青睐,但该方法由于过度依赖于DNA提取技术和PCR技术,DNA提取效率和PCR技术的缺陷必会影响检测结果的真实性,因此需要不断改进DNA提取技术与PCR技术,例如改进DNA的提取效率以及引物的特异性等,以此来增强RT-qPCR技术检测结果的可靠性。
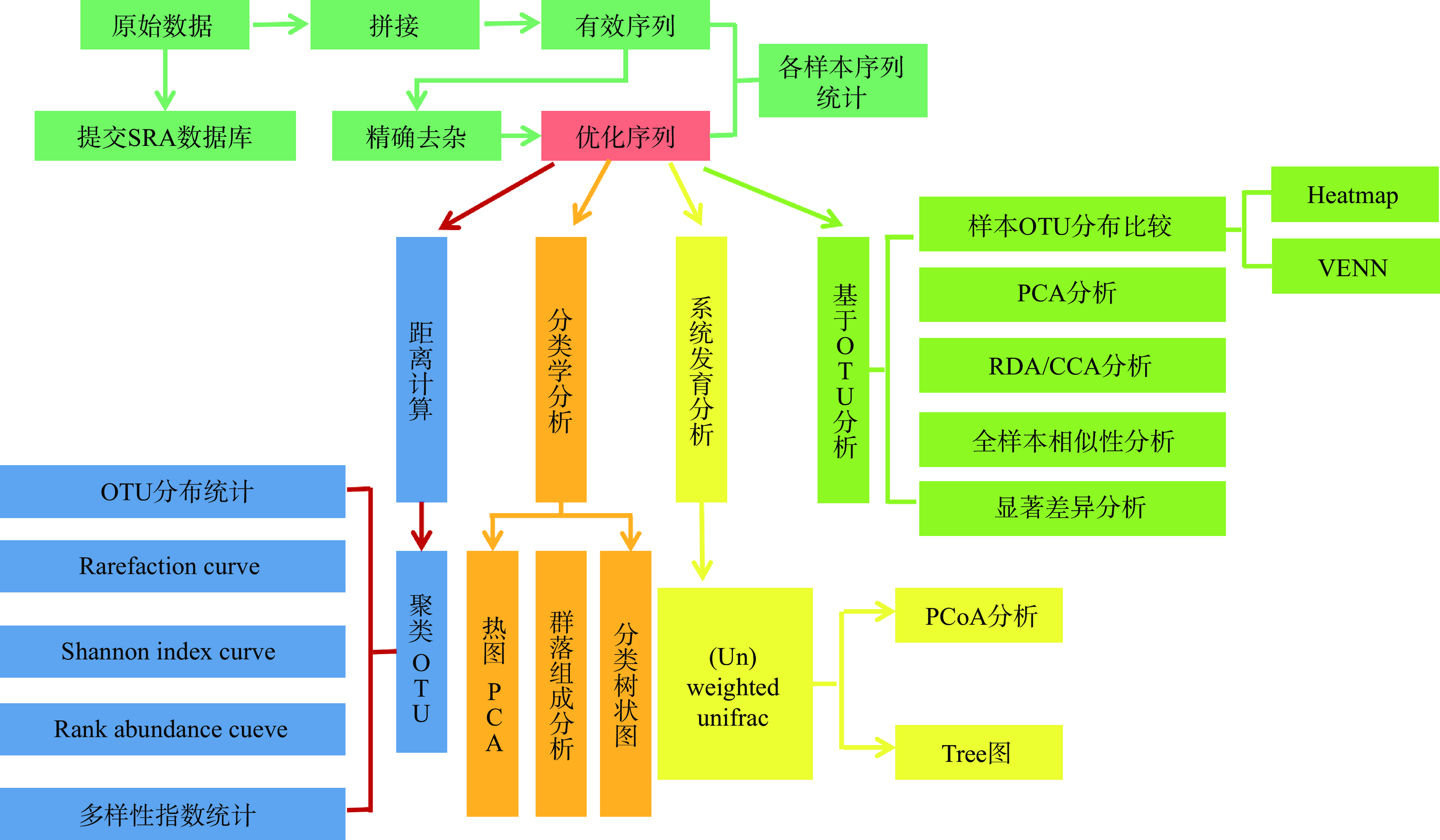
4. 高通量测序
高通量测序技术(High-throughput sequencing,HTS)又称为新一代测序技术(next generation sequencing,NGS),现已由第一代测序技术衍化到第三代测序技术,包括扩增子测序(amplicon sequencing)、宏基因组测序(metagenomics sequencing)、转录组测序(transcriptome sequencing)等。高通量测序技术的实验流程见图5[50]。高通量测序技术已被广泛地应用于环境微生物分子生态学研究中,但高通量测序技术需要依靠测序仪对整个微生物组的基因信息进行检测,导致其检测成本相对较高,同时对于16S rRNA测序来说,需要对样本进行PCR扩增,将会使结果产生一定的误差,对于宏基因组测序和宏转录组测序来说,需要具备雄厚的生物信息学知识,且最终只能得出微生物群落的相对丰度,并不能得出具体的数量。
4.1 扩增子测序技术
扩增子测序技术是一种基于PCR扩增微生物种群基因组特定区域并通过高通量测序研究样本中微生物组成及功能的方法。因其具有快速、简单、廉价的优点,被广泛应用于鉴定细菌和古菌的16S rRNA基因、真核生物的18S rRNA基因以及真菌的ITS区域当中。由于目前使用通用引物和二代测序平台,会使测序结果产生较大误差,导致生物功能信息的获取具有一定的局限性[51]。因此,结合其他微生物分子生态学研究方法来解决所获得生物功能信息受限的问题迫在眉睫。
扩增子测序技术,即先从样本中提取完整的DNA,随后利用通用引物扩增该“特定区域”基因并完成建库测序。周天慈等[52]利用高通量扩增子测序技术对中高温大曲中微生物的来源进行了分析,为优化制曲工艺,提升白酒品质提供理论支撑。Zhao等[53]利用16S rRNA扩增子测序分析了四川工业泡菜的细菌谱,发现了四川工业泡菜中的优势菌和产香菌。Kamimura等[54]利用16S rRNA扩增子测序分析了手工奶酪中的细菌多样性,显示发酵剂在微生物群塑造方面具有很强的相关性。扩增子测序技术在剖析微生物群落方面具有快速且成本较低的优点,但由于PCR扩增和引物亲和力的差异导致检测结果产生偏差,同时分辨率较低,仅区分到属级。与之相比,宏基因组在物种层面上进行鉴定,能够研究目前存在的微生物的功能潜力,并可能揭示未知物种。
4.2 宏基因组
宏基因组即环境中全部微生物基因的总和。它主要针对环境样品中微生物群落的遗传物质进行研究,可充分发掘未培养和未鉴定微生物的基因多样性与生化反应[55]。
近年来,以高通量测序为基础的宏基因组学现已被广泛应用于酒曲等其他食品微生物组研究中,为认识和筛选功能微生物提供了重要指导[56]。王正等[57]采用多组学联用技术探究了谷物蛋白对白酒发酵过程中微生物群落及其代谢多样性的影响,为提高白酒发酵的可控性及质量提供了依据。Wang等[58]利用高通量测序解析了酒曲中功能微生物菌群的组成情况。Lu等[59]基于功能基因组学的分析,揭示了中国茅台风味酵母MT1的潜在特征。课题组前期对来自重庆仙女山草原的黄牛粪便样品进行宏基因组测序,并通过比对和筛选得到7个公认的木聚糖异构酶基因,在大肠杆菌和酿酒酵母中进行异源表达证明了其酶活力[60]。
宏基因组学技术相对于传统培养和分子指纹图谱在获得数据量和可操作性方面有了很大进步,但仍不够完善,尚存在着诸多的问题:现有的算法无法完全利用庞大的测序数据而造成的浪费;DNA经剪切后再拼接难以控制完整片段的阈值而造成的误差等[61]。目前,研究人员发现“浅宏基因组”可以完美解决这一问题,该方法测序深度相对较低,但物种分辨率并没有低于一般宏基因组,而且具有更高的性价比[62]。
4.3 宏转录组
转录组学是指一门在整体水平上研究细胞中基因的表达情况及转录调控规律的学科,目前主要存在三种转录组处理技术:RT-PCR、微阵列和下一代RNA测序。
随着测序技术的发展,宏转录组学的方法在环境微生物的研究中被广泛应用,包括土壤、湖泊和海洋等生态系统。Bei等[63]通过宏基因组和宏转录组阐明了意大利改良稻田中Ignavibacteria的遗传潜力和功能活性。Mojib等[64]使用原位宏转录组学方法揭示了红海中代谢活跃的中型浮游动物群落的功能基因组组成。Rippin等[65]分别使用宏转录组和形态学鉴定生物土壤结皮中群落结构,显示出宏转录组高丰富度的优点。这些研究表明,与单一方法相比,多种技术的结合才能揭示复杂群落的更高属丰富度。
如果说宏基因组可以表明微生物群“能做什么”,那么宏转录组则可以说明微生物“想做什么”[66]。但是,宏转录组学与宏基因组学一样,需要依赖于测序技术和生物信息学分析,那么势必会导致以下结果[67]:基因组大小出现“缩水”现象;浪费部分测序的数据;宏转录组测序数据量较大,且不同的算法可能结果差异甚大;对低丰度基因的检测较困难;费用较高,并使结果的可靠性和再现性降低。
5. 功能基因组
功能基因组学常见的分析方法有基因芯片和RNA测序等技术,又称为后基因组学,是指基于基因组序列信息,利用各种组学技术,在系统水平上将基因组序列与基因功能(包括基因网络)以及表型有机联系起来,最终揭示自然界中生物系统不同水平的功能的科学。
5.1 基因芯片
近年来,基因芯片的发展及其在环境DNA杂交分析中的应用,极大地促进了对环境微生物种群多样性和活性的分析。在这种方法中,样本DNA通常是在基于PCR扩增之后,荧光标记并与基因芯片接触。在基因芯片上,多达数以万计的核苷酸探针,无论是由16S rRNA基因片段或功能基因片段组成[68],都被置于一个密集阵列中。样本中与芯片上的探针同源的基因,将会通过基因组杂交来获得其同源对应基因的位置。经过杂交,芯片上的信号被数字化分析。这样可以在高通量下获得系统发育多样性和群落组成以及功能位置的信息。
基因芯片适用于不同环境样本群落组成及功能的分析,Taguchi等[69]开发了一种不需要对目标核酸片段进行荧光标记、扩增或洗涤的新型基因芯片系统,与传统的依赖PCR的基因芯片相比,速度明显更快。Shin等[70]使用基因芯片和BacT/Alert培养系统分别检测含抗生素样品中的大肠杆菌和肺炎克雷伯氏菌,基因芯片在抗菌治疗后约4~24 h获得真阳性结果,其中基于培养的方法未能检测到病原菌,证实基因芯片可用于有效检测适当使用抗生素患者临床样本中的病原菌。Sohrabi等[71]使用基因芯片和组织学来检查肉类的成分和安全性,提高牛肉行业的监测能力。目前基因芯片存在探针设计困难和相似基因互相杂交等一系列问题,因此探针开发、杂交质量和数据评价是合理应用DNA微阵列研究环境微生物区系的关键步骤。
5.2 RNA测序
RNA测序即转录组测序技术,就是用高通量测序技术进行测序分析,反映出mRNA、smallRNA、noncodingRNA等或者其中一部分的表达水平。
在过去的二十年中,RNA测序二技术迅速发展,并成为了在转录组水平上分析差异基因表达或mRNA可变剪切的不可缺少的工具。Tan等[72]开发了一种通过标准RNA测序识别RNA修饰的ModTect统计框架,表明转录组失调与癌症进展和生存结果相关,可用于发现患者RNA修饰与临床结果之间的关联。Kim等[73]对35个果园的梨树叶片样本进行了RNA测序,基于转录组数据构建了三个完整的基因组,并进行了系统发育分析,确定苹果茎沟病毒(ASGV)和苹果茎凹坑病毒(ASPV)为韩国主要梨品种“Singo”的优势病毒。目前,RNA测序可以在单细胞水平上对细胞群进行特异性分析,揭示每种细胞类型的独特变化,但RNA测序对低丰度转录本的检测性能有待提高,可通过技术改进和优化分析方法解决。
6. 展望
微生物在全球尺度上调控着生物地球化学循环过程,影响生态系统的功能。理解生态系统功能,从群落和单细胞水平揭示自然环境中微生物的种类和功能、微生物之间以及微生物与环境因子之间的相互作用是必不可少的。
分子生物学方法的发展和进步为环境微生物的研究提供了方便,但是不同方法各有其内在的局限性,在方法的应用上需根据研究具体情况进行选择。例如,DGGE、T-RFLP、FISH、RT-qPCR、高通量测序和宏组学等研究方法均是近些年快速发展的方法,与纯培养法、Biolog微平板法和PLFA图谱分析等传统方法相比,微生物多样性分析的广度、深度和分辨率均有显著提高,显示出了其前所未有的优势和应用前景。
然而,这些方法本身仍存在一些问题:
a. 基于DNA序列的方法,由于不能区分活细胞和死细胞,可能会导致假阳性结果[74],如RT-qPCR在扩增过程中,以死细胞或损伤细胞的 DNA 作为PCR模板,将会增大检测结果[75],为克服这一缺陷,有研究发现脱氧胆酸钠(SD)和单叠丙酸丙啶(PMA)与 RT-PCR 技术相结合,可有效消除死亡细胞和损伤细胞对PCR扩增的影响[76]。
b. 基于PCR的方法,由于引物的选择以及 PCR本身的问题易对微生物的理解带来偏差,可利用Bellerophon[77]、Uchime[78]等软件并通过生物信息学分析移除假序列。
c. 高通量测序[79]得到的多为几百个碱基或者更短的序列,组成完整的基因有一定困难;即使获得整条序列,由于很多基因的功能还不清楚,很难获得由序列到功能的准确图谱。
未来的工作中,宏组学和单细胞水平研究的方法将有助于揭示自然环境中微生物整个群落的分类、功能信息以及丰度较低但发挥重要功能作用的微生物种群,然而,这并不代表着传统方法的研究应被终止,因为宏组学方法的研究依赖于培养技术所产生的参考数据库。同时,单细胞水平研究方法中的拉曼光谱-荧光原位杂交和纳米二次离子质谱-荧光原位杂交为环境中微生物的单细胞生态学和代谢潜力的研究开辟了一个新的途径。随着测序技术和单细胞水平研究技术等微生物分子生物学方法的不断改进及数据分析工具的不断发展,对环境中微生物群落组成、微生物之间的相互作用的认识以及对生态系统的功能的理解也在不断加深,对进一步深入了解微生物群落乃至微生态系统具有重要意义。
-
![]()
图 1 变性梯度凝胶电泳(DGGE)原理图
Figure 1. Schematic diagram of denaturing gradient gel electrophoresis (DGGE)
![]()
图 2 末端限制性长度多态性原理图
Figure 2. Schematic diagram of terminal restricted length polymorphism
![]()
图 4 实时荧光定量PCR原理图
Figure 4. Principle diagram of real-time fluorescence quantitative PCR
表 1 微生物分子生态学研究方法比较
Table 1 Comparison of research methods of microbial molecular ecology
类别 代表技术 优点 缺点 应用
案例
传
统
方
法分离培养法 简便易行 只能分离有限种类的微生物 [7] Biolog微平板法 可重复且容易使用,可以产生大量
反映群落代谢特征的数据只能检测到一些代谢活性高和可培养的细菌,不能反映真菌和生长缓慢的细菌,检测结果会受到接种密度的干扰 [8] PLFA图谱分析 简单快速,不需要特殊的设备,
对菌群环境变化敏感[9]分类分辨率差,结果受提取方法和菌群pH动态变化的影响,会产生一定的误差 [10]
D
N
A
指
纹
图
谱变性梯度凝胶电泳(DGGE) 简单,无放射性,相对无毒,检测率
和特异性高,杂合子检测率高分析高GC含量和高长度片段困难,分析费时费力且费用高,PCR引物不同会引起一定的误差 [11] 末端限制性长度多态性(T-RFLP) 可产生大量重复且分辨率高的结果,数据具体化,易实现自动化检测,操作简便,灵敏度高 无法区分近缘种微生物,不能确定各类微生物的绝对含量,片段长度范围的确定、噪声峰的去除及大小相近的片段合并都会对结果造成影响 [12]
荧光
标记荧光原位杂交技术(FISH) 经济、安全且探针稳定,实验周期短、特异性好、定位准确,可定位1 kb长度的DNA等 杂交效率低且检测灵敏度不高 [13] 实时荧光定量PCR 快速精确,解决了不可培养和未知微生物的定量问题,可以达到种的水平 无法得到整个微生物的群落的信息,实验难度大,
对实验结果有影响[14]
高
通
量
测
序
扩增子测序测序片段短,无需构建基因文库,高输出量和高解析度且测序结果覆盖整个微生物群落的信息 仪器设备昂贵,测序成本高,只能获得样品中微生物的相对丰度,无法确定样品中微生物的具体数量,对于16S rRNA测序来说,需要对样本进行PCR扩增,将会使结果
产生一定的误差[15]
宏基因组学高分辨率,没有因使用引物和PCR而产生的误差,直接对全部DNA进行测序,节省了
许多不必要的步骤[16]
需要一定的生物信息学知识储备和分析设备[17]
宏转录组学可以检测完整的转录组(编码和非编码RNA),可以检测RNA序列的变体和异构体,不需要微生物
基因组的知识储备样品制备步骤比较繁杂且样本的数量较少,需要经过
复杂的数据分析才可得到最终的实验结果[18] 功能
基因组学基因芯片 灵敏度高,可直接对高通量序列信息进行分析 低同源序列的交叉杂交导致分析结果产生偏差 [19] RNA测序 可以在单细胞水平上对细胞群进行特异性分析 对低丰度转录本的检测性能有待提高 [20]  下载: 导出CSV
下载: 导出CSV
-
[1] THOMPSON L R, SANDERS J G, MCDONALD D, et al. A communal catalogue reveals earth’s multiscale microbial diversity[J]. Nature,2017,551(7681):457−463. doi: 10.1038/nature24621
[2] WANG X, DU H, XU Y. Source tracking of prokaryotic communities in fermented grain of chinese strong-flavor liquor[J]. International Journal of Food Microbiology,2017,244:27−35. doi: 10.1016/j.ijfoodmicro.2016.12.018
[3] 王柏文, 吴群, 徐岩, 等. 中国白酒酒曲微生物组研究进展及趋势[J]. 微生物学通报,2021,48(5):1737−1746. [WANG B W, WU Q, XU Y, et al. Recent advances and perspectives in study of microbiome in chinese jiuqu starter[J]. Microbiology China,2021,48(5):1737−1746. doi: 10.13344/j.microbiol.china.200650 WANG B W, WU Q, XU Y, et al. Recent advances and perspectives in study of microbiome in chinese jiuqu starter[J]. Microbiology China, 2021, 48(05): 1737-1746. doi: 10.13344/j.microbiol.china.200650
[4] STACY A, MCNALLY L, DARCH S E, et al. The biogeography of polymicrobial infection[J]. Nature Reviews Microbiology,2016,14:93−105. doi: 10.1038/nrmicro.2015.8
[5] MIZRAHI I, WALLACE R J, MORAÎS S. The rumen microbiome: balancing food security and environmental impacts[J]. Nature Reviews Microbiology,2021,19:553−566. doi: 10.1038/s41579-021-00543-6
[6] LU M, REN Y L, WANG S J, et al. Contribution of soil variables to bacterial community composition following land use change in napahai plateau wetlands[J]. Journal of Environmental Management,2019,246(Sep.15):77−84.
[7] 冯明谦, 刘德明. 滚筒式高温堆肥中微生物种类数量的研究[J]. 中国环境科学,1999(6):490−492. [FENG M Q, LIU D M. Study on microbe species for high-temperature composting of horizontal cylinder[J]. China Environmental Science,1999(6):490−492. doi: 10.3321/j.issn:1000-6923.1999.06.003 FENG M Q, LIU D M. Study on microbe species for high-temperature composting of horizontal cylinder[J]. China Environmental Science, 1999(06): 490-492. doi: 10.3321/j.issn:1000-6923.1999.06.003
[8] SHANG Q, YANG G, WANG Y, et al. Illumina-based analysis of the rhizosphere microbial communities associated with healthy and wilted lanzhou lily (Lilium davidii var. unicolor)plants grown in the fifield[J]. World Journal of Microbiology & Biotechnology,2016,32(6):1−15.
[9] WATZINGER A. Microbial phospholipid biomarkers and stable isotope methods help reveal soil functions[J]. Soil Biology and Biochemistry,2015,86:98−107. doi: 10.1016/j.soilbio.2015.03.019
[10] JSB A, BV B, JM A, et al. Combined extraction method for metabolomic and PLFA analysis of soil[J]. Applied Soil Ecology,2019,135:129−136. doi: 10.1016/j.apsoil.2018.11.012
[11] LIANG H, CHEN H, JI C, et al. Dynamic and functional characteristics of predominant species in industrial paocai as revealed by combined DGGE and metagenomic sequencing[J]. Frontiers in Microbiology,2018:9.
[12] SINGH A, MÛLLER B, SCHNÛRER A. Profiling temporal dynamics of acetogenic communities in anaerobic digesters using next-generation sequencing and T-RFLP[J]. Scientific Reports, 2021.
[13] SUI C P, CABOT J M, MACKA M, et al. Isotachophoretic fluorescence in situ hybridization of intact bacterial cells[J]. Anal Chem, 2017.
[14] SALES M L, DALL'AGNOL M, OLIVEIRA AMD , et al. RT-qPCR for the diagnosis of the vesiculovirus cocal virus[J]. Archives of Virology, 2020.
[15] JONES B M, KUSTKA A B. A quantitative SMRT cell sequencing method for ribosomal amplicons[J]. Journal of Microbiological Methods,2017,135(Complete):77−84.
[16] GEISEN S, BONKOWSKI M. Methodological advances to study the diversity of soil protists and their functioning in soil food webs[J]. Applied Soil Ecology,2018,123:328−333. doi: 10.1016/j.apsoil.2017.05.021
[17] LAMAS A, REGAL P, VÀZQUEZ B, et al. Transcriptomics: A powerful tool to evaluate the behavior of foodborne pathogens in the food production chain[J]. Food Research International,2019,125:108543. doi: 10.1016/j.foodres.2019.108543
[18] MAUCHLINE T H, HAYAT R, CLARK I M, et al. Old meets new: most probable number validation of metagenomic and metatranscriptomic datasets in soil[J]. Letters in Applied Microbiology, 2018.
[19] MENG X, YU Y, GONG P, et al. An integrated droplet digital PCR gene chip for absolute quantification of nucleic acid[J]. Microfluidics and Nanofluidics, 2021, 25(7): 1-9.
[20] LANDOLT L, MARTI H P, BEISLAND C, et al. RNA extraction for RNA sequencing of archival renal tissues[J]. Scandinavian Journal of Clinical & Laboratory Investigation,2016:426−434.
[21] NYKYRI J, HERRMANN A M, HÅKANSSON S. Isothermal microcalorimetry for thermal viable count of microorganisms in pure cultures and stabilized formulations[J]. Bmc Microbiology, 2019, 19(1):65.
[22] EI-LIETHY M A, HEMDAN B A, EI-TAWEEL G E. Phenotyping using semi-automated BIOLOG and conventional PCR for identification of bacillus isolated from biofilm of sink drainage pipes[J]. Acta Ecologica Sinica,2018,38(5):334−338. doi: 10.1016/j.chnaes.2018.01.011
[23] MORGAN M C, BOYETTE M, GOFORTH C, et al. Comparison of the Biolog OmniLog Identification System and 16S ribosomal RNA gene sequencing for accuracy in identification of atypical bacteria of clinical origin[J]. Journal of Microbiological Methods,2009,79(3):336−343. doi: 10.1016/j.mimet.2009.10.005
[24] GUANG H W, JUN J L, XIAO N Q, et al. Effects of fertilization on bacterial community structure and function in a black soil of dehui region estimated by Biolog and PCR-DGGE methods[J]. Acta Ecologica Sinica,2008,28(1):220−226. doi: 10.1016/S1872-2032(08)60023-2
[25] CHEN H, ZHAO X, LIN Q, et al. Using a combination of PLFA and DNA-based sequencing analyses to detect shifts in the soil microbial community composition after a simulated spring precipitation in a semi-arid grassland in China[J]. Science of The Total Environment,2019,657:1237−1245. doi: 10.1016/j.scitotenv.2018.12.126
[26] ORWIN K H, DICKIE I A, HOLDAWAY R, et al. A comparison of the ability of PLFA and 16S rRNA gene metabarcoding to resolve soil community change and predict ecosystem functions[J]. Soil Biology and Biochemistry,2018,117:27−35. doi: 10.1016/j.soilbio.2017.10.036
[27] 赵妍, 刘顺杰, 张亚茹, 等. 微生物多样性分析技术应用于食用菌发酵培养料分析的进展[J]. 食用菌学报,2019,26(3):148−156. [ZHAO Y, LIU S J, ZHANG Y R, et al. Advances in application of microbial diversity analysis techniques on analyzing edible fungi fermented substrates[J]. Acta Edulis Fungi,2019,26(3):148−156. doi: 10.16488/j.cnki.1005-9873.2019.03.018 ZHAO Y, LIU S J, ZHANG Y R, et al. Advances in application of microbial diversity analysis techniques on analyzing edible fungi fermented substrates[J]. Acta Edulis Fungi, 2019, 26(3): 148-156. doi: 10.16488/j.cnki.1005-9873.2019.03.018
[28] LI Z H, RUI J P, LI X Z, et al. Bacterial community succession and metabolite changes during doubanjiang-meju fermentation, a Chinese traditional fermented broad bean (Vicia faba L. ) paste[J]. Food Chemistry,2017,218:534−542. doi: 10.1016/j.foodchem.2016.09.104
[29] COSTA M, WEESE J S. Methods and basic concepts for microbiota assessment[J]. The Veterinary Journal,2019,249:10−15. doi: 10.1016/j.tvjl.2019.05.005
[30] MUYZER G, SMALLA K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology[J]. Antonie Van Leeuwenhoek,1998,73(1):127−141. doi: 10.1023/A:1000669317571
[31] XIONG Z Q, LI Y Y, XIANG Y W, et al. Short communication: dynamic changes in bacterial diversity during the production of powdered infant formula by PCR-DGGE and high-throughput sequencing[J]. Journal of Dairy Science,2020,103(7):5972−5977. doi: 10.3168/jds.2019-18064
[32] CHAHORM K, PRAKITCHAIWATTANA C. Application of Reverse Transcriptase-PCR-DGGE as a rapid method for routine determination of Vibrio spp. in foods[J]. International Journal of Food Microbiology,2017:264.
[33] BO B, KIM S A, HAN N S. Bacterial and fungal diversity in Laphet, traditional fermented tea leaves in myanmar, analyzed by culturing, DNA amplicon-based sequencing, and PCR-DGGE methods[J]. International Journal of Food Microbiology,2020,320:108508. doi: 10.1016/j.ijfoodmicro.2020.108508
[34] MARSH T L . Terminal restriction fragment length polymorphism (T-RFLP): An emerging method for characterizing diversity among homologous populations of amplification products[J]. Current Opinion in Microbiology, 1999, 2(3): 323-327.
[35] LÓPEZ A C, ALIPPI A M. Feasibility of using RFLP of PCR-amplified 16S rRNA gene(s) for rapid differentiation of isolates of aerobic spore-forming bacteria from honey[J]. Journal of Microbiological Methods,2019,165:105690. doi: 10.1016/j.mimet.2019.105690
[36] ROSA N M, AGNOLETTI F, LOLLAI S, et al. Comparison of PCR-RFLP, API® 20 Strep and MALDI-TOF MS for identification of Streptococcus spp. collected from sheep and goat milk samples[J]. Small Ruminant Research,2019,180:35−40. doi: 10.1016/j.smallrumres.2019.09.023
[37] 李甜甜, 胡泓, 王金爽, 等. 湿地土壤微生物群落结构与多样性分析方法研究进展[J]. 土壤通报,2016,47(3):758−762. [LI T T, HU H, WANG J S, et al. Progress in research methods of soil microbial structure and diversity in wetlands[J]. Chinese Journal of Soil Science,2016,47(3):758−762. doi: 10.19336/j.cnki.trtb.2016.03.38 LI T T, HU H, WANG J S, et al. Progress in Research Methods of Soil Microbial Structure and Diversity in Wetlands. [J]. Chinese Journal of Soil Science, 2016, 47(3): 758-762. doi: 10.19336/j.cnki.trtb.2016.03.38
[38] HONG P, YAO X, CHEN W, et al. Dissecting complicated viral spreading of enterovirus 71 using in situ bioorthogonal fluorescent labeling[J]. Biomaterials,2018,181:199−209. doi: 10.1016/j.biomaterials.2018.07.061
[39] HUBER D, VOITH VON VOITHENBERG L, KAIGALA G V. Fluorescence in situ hybridization (FISH): History, limitations and what to expect from micro-scale FISH?[J]. Micro and Nano Engineering,2018,1:15−24. doi: 10.1016/j.mne.2018.10.006
[40] CUI C, SHU W, LI P. Fluorescence in situ hybridization: Cell-based genetic diagnostic and research applications[J]. Frontiers in Cell and Developmental Biology,2016:4.
[41] 宋伟凤, 李明聪, 高峥. 环境中微生物原位检测方法研究进展[J]. 生物技术通报,2017,33(10):26−32. [SONG W F, LI M C, GAO Z. Research progress onin situ detection methods of microorganisms[J]. Biotechnology Bulletin,2017,33(10):26−32. doi: 10.13560/j.cnki.biotech.bull.1985.2017-0550 SONG W F, LI M C, GAO Z. Research Progress on in situ Detection Methods of Microorganisms[J]. Biotechnology Bulletin, 2017, 33(10): 26-32. doi: 10.13560/j.cnki.biotech.bull.1985.2017-0550
[42] CHU Y H, HARDIN H, ZHANG R R, et al. In situ hybridization: Introduction to techniques, applications and pitfalls in the performance and interpretation of assays[J]. Seminars in Diagnostic Pathology,2019,36(5):336−341. doi: 10.1053/j.semdp.2019.06.004
[43] ZHANG B, MAIMAITI Y, LIU C, et al. Direct detection of Staphylococcus aureus in positive blood cultures through molecular beacon-based fluorescence in situ hybridization[J]. Journal of Microbiological Methods,2019,159:34−41. doi: 10.1016/j.mimet.2019.02.007
[44] SALIMI G, MOUSAVI E, KIANI H. Efficiency of fluorescence in situ hybridization (FISH) method for the rapid detection of Salmonella in minced lamb meat: Method analysis and optimization[J]. Journal of Microbiological Methods,2020,175:105989. doi: 10.1016/j.mimet.2020.105989
[45] BALIGA S, MURPHY C, SHARON L, et al. Rapid method for detecting and differentiating Mycobacterium tuberculosis complex and non-tuberculous mycobacteria in sputum by fluorescence in situ hybridization with DNA probes[J]. International Journal of Infectious Diseases,2018,75:1−7. doi: 10.1016/j.ijid.2018.07.011
[46] TANG Y, ZOU B, WANG R, et al. Multiplex-invasive reaction-assisted qPCR for quantitatively detecting the abundance of EGFR exon 19 deletions in cfDNA[J]. Analytical Methods,2020:12.
[47] BAHLINGER E, DORN-IN S, BEINDORF P M, et al. Development of two specific multiplex qPCRs to determine amounts of Pseudomonas, Enterobacteriaceae, Brochothrix thermosphacta andStaphylococcus in meat and heat-treated meat products[J]. International Journal of Food Microbiology,2020:337.
[48] PANCZA B, SZATHMÁRY M, GYURJÁN I, et al. A rapid and efficient DNA isolation method for qPCR-based detection of pathogenic and spoilage bacteria in milk[J]. Food Control,2021:108236.
[49] BROTONS P, PEREZ-ARGÜELLO A, LAUNES C, et al. Validation and implementation of a direct RT-qPCR method for rapid screening of SARS-CoV-2 infection by using non-invasive saliva samples[J]. Journals & Books,2021,110:363−370.
[50] REON B J, DUTTA A. Biological processes discovered by high-throughput sequencing[J]. American Journal of Pathology,2016:722−732.
[51] TREMBLAY J, SINGH K, FERN A, et al. Primer and platform effects on 16S rRNA tag sequencing[J]. Frontiers in Microbiology,2015,6:771.
[52] 周天慈, 何宏魁, 徐岩, 等. 基于高通量扩增子测序技术解析中高温大曲微生物来源[J]. 食品与发酵工业, 2021, 47(16): 66−71. ZHOU T C, HE H K, XU Y, et al. Exploring the source of microbiota in medium-high temperature daqu based on high-throughput amplicon sequencing[J]. Food and Fermentation Industries, 2021, 47(16): 66−71.
[53] ZHAO Y J, WEI W L, TANG L, et al. Characterization of aroma and bacteria profiles of Sichuan industrial paocai by HS-SPME-GC-O-MS and 16S rRNA amplicon sequencing[J]. Food Research International, 2021(149).
[54] KAMIMURA B A, CABRAL L, NORONHA M F, et al. Amplicon sequencing reveals the bacterial diversity in milk, dairy premises and Serra da Canastra artisanal cheeses produced by three different farms[J]. Food Microbiology,2020,89(Aug.):103453.1−103453.12.
[55] BLAMEY J M, FISCHER F, MEYER H P, et al. Enzymatic biocatalysis in chemical transformations: A promising and emerging field in green chemistry practice-ScienceDierct[J]. Biotechnology of Microbial Enzymes,2017:347−403.
[56] HUANG Y H, YI Z L, JIN Y L, et al. Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese Nong-flavor liquor starter[J]. Frontiers in Microbiology,2017,8:1747. doi: 10.3389/fmicb.2017.01747
[57] 王正, 吴群, 徐岩, 等. 谷物蛋白对白酒发酵过程中微生物群落及其代谢多样性的调控[J]. 微生物学通报,2021(48):4167−4177. [WANG Z, WU Q, XU Y, et al. The regulation of grain protein on the microbial community and metabolic diversity in the process of liquor fermentation[J]. Microbiology China,2021(48):4167−4177. doi: 10.13344/j.microbiol.china.210228 WANG Z, WU Q, XU Y, et al. The regulation of grain protein on the microbial community and metabolic diversity in the process of liquor fermentation[J]. Microbiology China, 2021, 1-10. doi: 10.13344/j.microbiol.china.210228
[58] WANG B W, WU Q, XU Y, et al. Specific volumetric weight-driven shift in microbiota compositions with saccharifying activity change in starter for chinese baijiu fermentation[J]. Frontiers in Microbiology,2018,9:2349. doi: 10.3389/fmicb.2018.02349
[59] LU X W, WU Q, XU Y, et al. Genomic and transcriptomic analyses of the Chinese Maotai-flavored liquor yeast MT1 revealed its unique multi-carbon co-utilization[J]. Bmc Genomics,2015,16(1):1−14. doi: 10.1186/1471-2164-16-1
[60] TANG R Q, YE P L, ALPER H S, et al. Identification and characterization of novel xylose isomerases from a Bos taurus fecal metagenome[J]. Applied Microbiology and Biotechnology,2019,103(11):1−13.
[61] 王禄禄, 王立志, 周美丽. 宏基因组学技术在反刍动物瘤胃微生态系统上的应用研究进展[J]. 中国微生态学杂志,2017,29(2):223−228. [WANG L L, WANG L Z, ZHOU M L. Application of metagenomics technology on the microecological system in rumen of ruminant: Research progress[J]. Chinese Journal of Microbiology,2017,29(2):223−228. doi: 10.13381/j.cnki.cjm.201702028 WANG L L, WANG L Z, ZHOU M L. Application of metagenomics technology on the microecological system in rumen of ruminant: Research progress[J]. Chinese Journal of Microbiology, 2017, 29(02): 223-228. doi: 10.13381/j.cnki.cjm.201702028
[62] ALI A, CHRISTOPHERSEN C T, KEELAN J A. Vaginal microbial profiling in a preterm birth high-risk cohort using shallow shotgun metagenomics[J]. Microbiology Australia, 2021, 42(2): 69-74.
[63] BEI Q H, PENG J J, LIESACK W. Shedding light on the functional role of the Ignavibacteria in Italian rice field soil: A meta-genomic/transcriptomic analysis[J]. Soil Biology and Biochemistry,2021:163.
[64] MOJIB N, THIMMA M, KUMARAN M, et al. Comparative metatranscriptomics reveals decline of a neustonic planktonic population[J]. Limnology and Oceanography,2017,62:299−310. doi: 10.1002/lno.10395
[65] RIPPIN M, BORCHHARDT N, WILLIAMS L, et al. Genus richness of microalgae and cyanobacteria in biological soil crusts from svalbard and livingston island: morphological versus molecular approaches[J]. Polar Biology,2018,41(5):1−15.
[66] LI F, NEVES ALA, GHOSHAL B, et al. Symposium review: mining metagenomic and metatranscriptomic data for clues about microbial metabolic functions in ruminants[J]. Journal of Dairy Science,2018,101(6):5605−5618. doi: 10.3168/jds.2017-13356
[67] 雷忠华, 陈聪聪, 陈谷. 基于宏基因组和宏转录组的发酵食品微生物研究进展[J]. 食品科学,2018,39(3):330−337. [LEI Z H, CHEN C C, CHEN G. Metagenomic and metatranscriptomic analysis of microbiota in fermented foods: Review of recent advances[J]. Food Science,2018,39(3):330−337. doi: 10.7506/spkx1002-6630-201803049 LEI Z H, CHEN C C, CHEN G. Metagenomic and metatranscriptomic analysis of microbiota in fermented foods: review of recent advances[J]. Food Science, 2018, 39(3): 330-337. doi: 10.7506/spkx1002-6630-201803049
[68] DESANTIS T Z, BRODIE E L, MOBERG J P, et al. High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment[J]. Microbial Ecology, 2007, 53(3): 371−383.
[69] TAGUCHI T, ISHIKAWA M, ICHIKAWA M, et al. Amplification-free detection of bacterial genes using a signaling probe-based DNA microarray [J]. Biosensors and Bioelectronics, 2021, 194(113659).
[70] SHIN S Y, KIM D M, JO Y, et al. DNA Microarray-based detection of bacteria in samples containing antibiotics: Effect of antibiotics on the performance of pathogen detection assays[J]. Biotechnology and Bioprocess Engineering,2021,26(3):447−455. doi: 10.1007/s12257-020-0342-9
[71] SOHRABI H, CANNIZZO F T, PREGEL P, et al. Tissue and species identification in minced meat and meat products from Italian commercial markets by DNA microarray and histological approach [J].Veterinaria Italiana, 2020, 56(2): 77−85.
[72] TAN K T, DING L W, WU C S, et al. Repurposing RNA sequencing for discovery of RNA modifications in clinical cohorts[J]. Science Advances,2021,7(32):1−26.
[73] KIM N Y, LEE H J, KIM H S, et al. Identification of plant viruses infecting pear using RNA sequencing[J]The Plant Pathology Journal, 2021, 37(3): 258−267.
[74] REN Q D, WEI F Q, YUAN C, et al. The effects of removing dead bacteria by propidium monoazide on the profile of salivary microbiome[J]. BMC Oral Health,2021,460:21.
[75] MIOTTO M, BARRETTA C, OSSAi S O, et al. Optimization of a propidium monoazide-qPCR method for Escherichia coli quantification in raw seafood[J]. International Journal of Food Microbiology,2019,318:108467.
[76] ZHAO S, ZHANG J Y, LI Z, et al. Enumeration of viable non-culturable vibrio cholerae using droplet digital PCR combined with propidium monoazide treatment[J]. Frontiers in Cellular and Infection Microbiology,2021,11:753078. doi: 10.3389/fcimb.2021.753078
[77] KERKVLIET J, ARTHUR D F, MICHIEL V W, et al. The Bellerophon pipeline, improving de novo transcriptomes and removing chimeras[J]. Ecology and Evolution, 2019, 9(18): 10513-10521.
[78] EDGAR R C, HAAS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011.
[79] FITZPATRICK A H, RUPNIK A, O'SHEA H, et al. High throughput sequencing for the detection and characterization of RNA viruses[J]. Frontiers in Microbiology,2021,12:621719. doi: 10.3389/fmicb.2021.621719
-
期刊类型引用(19)
1. 许津阁,郑卓琦,侯鹏颉,马高兴,熊彦娣,马壮,刘萌,赵靓,廖小军. 不同产地酱用卡宴辣椒原料品质评价. 食品工业科技. 2025(01): 317-332 .  本站查看
本站查看
2. 任朝辉,何建文,田怀志,田浩,廖卫琴. 基于主成分和聚类分析不同辣椒资源农艺和品质性状的综合评价. 中国瓜菜. 2025(02): 50-58 . 百度学术
3. 杨晶,沙迪昕,张月,麦迪乃·尤努斯,沙黑兰·尼亚孜,杨海燕,黄文书. 不同贮藏条件下干辣椒颜色劣变的主要途径. 食品研究与开发. 2024(04): 58-67 . 百度学术
4. 龙会英,张德. 干热区紫花苜蓿的生产性能和营养价值评价. 草业科学. 2024(01): 117-125 . 百度学术
5. 李莹,张娇,杨树辉,朱月,陈滕,汪祖华. 不同温度和气体微环境对遵义干辣椒贮藏品质的影响. 食品研究与开发. 2024(11): 80-88 . 百度学术
6. 裴艳婷,魏龙雪,李娜娜,白静,朱金英. 不同辣椒种质资源品质性状分析. 安徽农业科学. 2024(11): 27-31 . 百度学术
7. 马唯钦,赵牧其尔,孙鹏波,刘逸超,李子琪,贾玉山,格根图,王志军. 10个饲用燕麦品种在沿黄盐碱地区生产性能评价. 饲料研究. 2024(22): 139-144 . 百度学术
8. 廖卫琴,何建文,苟晓松,任朝辉,田浩. 不同辣椒种质资源果实中脂肪酸组成分析. 辣椒杂志. 2024(04): 13-18 . 百度学术
9. 年国芳,郭超男,徐建宗,周建中. 新疆制干辣椒品质综合评价及加工适宜性分析. 食品工业科技. 2023(04): 317-325 . 本站查看
10. 林巧,辛竹琳,孔令博,王晓梅,杨小薇,何微. 我国辣椒产业发展现状及育种应对措施. 中国农业大学学报. 2023(05): 82-95 . 百度学术
11. 詹磊,徐卓越,蓝国玮,钟庆玲,刘倩桐,陈佩. 基于主成分分析构建混合多糖凝胶品质综合评价模型. 现代食品科技. 2023(04): 214-223 . 百度学术
12. 杨创创,何建文,张正海,于海龙,冯锡刚,吴华茂,曹亚从,王立浩. 绥阳子弹头干椒风味品质分析. 辣椒杂志. 2023(01): 1-5+13 . 百度学术
13. 张新悦,连畅,宋文胜,郭涛,杜心宇,徐嘉悦,张新贵,孙志健,廖小军,赵靓. 新疆地区辣椒自然干制的关键节点品质分析. 食品工业科技. 2023(12): 90-101 . 本站查看
14. 屠大伟,翁盈秋,李青青,冯露萍,刘文俊. 火锅常用干辣椒品质及挥发性成分研究. 食品工业科技. 2023(16): 358-366 . 本站查看
15. 向家勇,杨莎,梁成亮,陈文超,李雪峰,欧立军,戴雄泽,马艳青,邹学校,张竹青. 鲜食青椒果实的品质性状分析与评价. 湖南农业大学学报(自然科学版). 2023(04): 436-441 . 百度学术
16. 杨芳,袁海彬,贾洪锋,邓凤琳,王珍妮. 基于气相色谱-离子迁移谱结合多元统计方法分析辣椒品种对辣椒油理化性质和风味物质的影响. 食品与发酵工业. 2023(19): 319-328 . 百度学术
17. 林素钦,马文婧,何新超,付桂明,钟剑,彭红,万茵. 不同油温对辣椒油风味和辣度的影响. 河南工业大学学报(自然科学版). 2023(05): 25-32 . 百度学术
18. 周鹏,杨娅,付文婷,王楠艺,彭世清,何建文. 贵州25个辣椒主栽品种品质分析与评价. 食品安全质量检测学报. 2023(21): 292-298 . 百度学术
19. 吴梓仟,周劲松,刘特元,蒋立文,刘洋,尹世鲜,荣智兴,陈欢. 基于HS-SPME-GC-MS分析不同卤制条件下卤制液香气差异. 食品工业科技. 2023(24): 311-318 . 本站查看
其他类型引用(7)
 下载:
下载:





计量
- 文章访问数: 363
- HTML全文浏览量: 668
- PDF下载量: 56
- 被引次数: 26
