


Response Surface Optimization of the QuEChERS Combined with UHPLC-MS/MS Method for Determination of Tetracyclines in Muscle of Channa argus
-
摘要: 为了评估乌鳢中四环素类药物的残留风险,建立了乌鳢肌肉中4种四环素类药物残留的快速、简单、廉价、高效、耐用和安全的前处理方法(Quick、Easy、Cheap、Effective、Rugged、Safe,QuEChER)。采用Box–Behnken design(BBD)响应面优化法,优化了提取和净化条件。样品经10 mL 1%甲酸酸化乙腈和2 mL 0.1 mol/L Na2EDTA-McIlvaine缓冲溶液提取,以150 mg PSA和200 mg C18进行净化,采用超高效液相色谱-串联质谱法(Ultra-high performance liquid chromatography-tandem mass spectrometry, UPLC-MS/MS)检测。结果表明,该方法在0.50~100 ng/mL浓度范围内线性良好(r≥0.999),检出限(Limit of Detection,LOD)和定量限(Limit of Quantitation,LOQ)分别为0.25~0.75 μg/kg和0.84~2.00 μg/kg。在乌鳢肌肉中四环素、土霉素、金霉素和多西环素的平均回收率在89.5%~1187%之间,相对标准偏差(RSD)在1.11%~8.88%之间。该方法简便、灵敏、准确,可用于乌鳢中四环素含量的快速测定。
-
关键词:
- QuEChERS /
- 超高效液相色谱-串联质谱 /
- 乌鳢 /
- 四环素类 /
- 风险评估
Abstract: A quick, easy, cheap, effective, rugged and safe pre-treatment method (QuEChERS) was established to evaluate the risk of tetracycline residues in the muscle of Channa argus. The Box-Behnken design (BBD) response surface optimization method was used to optimize the extraction and purification conditions. The samples were extracted with 10 mL acetonitrile (containing 1% formic acid) and 2 mL 0.1 mol/L Na2EDTA-Mcllvaine buffer solution, and the extracts were purified by 150 mg primary secondary amine(PSA)and 200 mg C18, and detected with Ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The result showed that the calibration curve was linear in the range of 0.50~100 ng/mL, with high correlation coefficients (r≥0.999). The limits of detection (LOD) and quantification (LOQ) were 0.25~0.75 μg/kg and 0.84~2.00 μg/kg, respectively. The average recoveries of tetracycline, oxytetracycline, chlortetracycline and doxycycline at three different spiked levels in the muscle of Channa argus were 89.5%~118%, with relative standard deviations (RSDs) of 1.11%~8.88%. This method is simple, sensitive and accurate, and can be used for the rapid quantitative determination of tetracyclines in Channa argus.-
Keywords:
- QuEChERS /
- UPLC-MS/MS /
- Channa argus /
- tetracycline /
- risk assessment
-
四环素类抗生素是一类对常见的革兰氏阳性菌、阴性菌均具有较强抗菌作用的广谱性抗生素,被广泛用于动物性疾病的预防治疗[1]。近年来随着水产养殖业的快速发展,集约化高密度养殖越来越普遍,导致养殖环境水体恶化,各种疾病频发。养殖者常超剂量长时间使用抗菌药物进行病害防治。四环素类抗菌药作为水产养殖中的限用药物,在淡水养殖中广泛使用。水产养殖中不合理使用的四环素类抗生素,会通过水生动物的吸收和代谢转移至水体环境,给环境安全带来风险[2−3]。通过水生动物的生物富集,药物在其组织中残留,随着人们食用水产品而进入人体,给人体健康带来危害[4−5]。因此,美国、欧盟、日本等制定了四环素类抗生素在不同食品中的最大残留限量(MRL),四环素类药物在可食动物中的MRL值分别为美国:2.0~12 mg/kg[6],欧盟:100~600 μg/kg[7],日本:0.2~1.2 mg/kg(总和)[8],我国规定四环素类总残留量不超过100 μg/kg[9]。
目前动物性食品中四环素类抗菌药的残留检测方法主要有薄层色谱法[10]、化学发光法[11]、免疫分析法[12−13]、高效液相色谱法[8,14−15]和液相色谱串联质谱法[16−17]。其前处理大多采用固相萃取(SPE)[18−21]、固相分散萃取(SPME)[19,22]、超声辅助提取(UAE)[15]、加速溶剂萃取(ASE)等[17,23]较为繁琐复杂的方法。Quick、Easy、Cheap、Effective、Rugged、Safe(QuEChERS)前处理方法是近年来迅速发展的一种快速前处理方法,具有快速、简单、低成本、耐用、可靠、安全和多类型化合物同时检测的特点,在药物残留检测中广泛应用[18-19,24-25],动物源食品中抗菌药物残留测定方面也有报道[23−24],采用QuEChERS前处理方法可以大大提高检测效率,获得更便捷和更准确的分析方法。目前四环素类药物残留检测方法大多采用固相萃取方法,操作繁琐且费时,本研究拟采用响应面法对影响方法准确性和精密度的前处理条件进行优化,有望建立一种简单快速且准确性和精密度较高的乌鳢肌肉中土霉素、金霉素、四环素和强力霉素的QuEChERS结合UPLC-MS/MS检测方法。并采用建立的方法测定广东主要养殖池塘的乌鳢样品中四环素残留量,分析其膳食风险,为乌鳢养殖中四环素类抗菌药物的风险评估及监管提供有效依据和技术支撑。
1. 材料与方法
1.1 材料与仪器
空白乌鳢样品 来自于中国水产科学研究院珠江水产研究所养殖基地,养殖过程未使用过四环素类药物;实际检测的乌鳢样品 均采集于广东主要养殖区域的养殖池塘;盐酸土霉素(OTC,纯度96.5%)、盐酸金霉素(CTC,纯度93%)、盐酸多西环素(DC,纯度98.7%)、盐酸四环素(TC,纯度96.5%) 标准品,德国Dr.Ehrenstorfer公司;氘代四环素(TC-D6,纯度>80%) 加拿大Toronto Research Chemicals(TRC)公司;二水合乙二胺四乙酸二钠(Na2EDTA·2H2O)、氯化钠(NaCl)、十二水合磷酸氢二钠(Na2HPO4·12H2O)、一水合柠檬酸(C6H8O7·H2O)、盐酸(HCl) 分析纯,广州化学试剂厂;甲酸、N-丙基乙二胺(PSA,40-63 μm)、中性氧化铝(Alumina-N,100-300 MESH)、弗罗里硅土(Florisil,150-250 μm) 德国CNW公司;乙酸铵 色谱纯,阿拉丁试剂(上海)有限公司;十八烷基键合硅胶吸附剂(C18,40-60 μm)、石墨化碳黑(GCB,120-400 MESH) 天津Agela公司;乙腈、甲醇 色谱纯,美国Burdick & Jackson公司;实验用水为超纯水。
1290-6470超高效液相色谱串联三重四极杆质谱仪,配有电喷雾离子源(ESI) 美国安捷伦公司;MS3旋涡混匀 德国IKA公司;Direct-Q3超纯水机 美国Millipore公司;TDL-5-A离心机 上海安亭公司;N-EVAP-12氮吹仪 美国Organomation公司;3k15高速离心机 美国Sigma公司;KQ5200型超声仪 昆山市超声仪器有限公司。
1.2 实验方法
1.2.1 标准溶液及试剂配制
标准溶液配制:准确称取适量的TC、OTC、CTC、DC和内标TC-D6 5种标准品,分别用甲醇溶解并定容,配制成质量浓度为100 μg/mL的单标储备液。准确移取四种单标标准储备液各1 mL用甲醇稀释并定容至10 mL,摇匀,配制成10 μg/mL的混合标准中间溶液,将100 μg/mL的TC-D6内标溶液用甲醇稀释成10 μg/mL标准中间液,于-18 ℃下避光保存备用。
0.1 mol/L Na2EDTA-McIlvaine(pH4.5)缓冲溶液配制[25]:分别准确称取37.2 g Na2EDTA·2H2O、10.9 g Na2HPO4·12H2O和12.9 g C6H8O7·H2O,用1.00 L超纯水充分溶解,加入适量盐酸调节pH为4.5,于4 ℃冰箱保存备用,使用前放至室温。
1.2.2 前处理方法
提取:准确称取2.00 g匀浆后的肌肉样品于50 mL具塞离心管中,加入2 mL 0.1 mol/L Na2EDTA-McIlvaine(pH4.5)缓冲溶液,涡旋混匀后加入10 mL 1%甲酸酸化乙腈和2.0 g 氯化钠,涡旋振荡2 min,5000 r/min离心5 min,上清液转移至50 mL离心管中待净化。
净化:提取液中加入500 mg无水硫酸钠、150 mg PSA和200 mg C18,涡旋1 min,5000 r/min离心5 min,将上清液转移至50 mL玻璃管中于45 °C下氮气吹干,然后用1 mL 15%甲醇水充分溶解后,10000 r/min离心5 min,过0.22 μm微孔滤膜,待UHPLC-MS/MS检测。
1.2.3 色谱条件
色谱柱:Agilent Zorbax SB-C18(2.1 mm×50 mm,18 µm);流动相A为0.5%甲酸水,B为甲醇;流速:0.40 mL/ min;柱温:30 ℃;进样体积:5 µL,梯度洗脱程序列于表1。
表 1 梯度洗脱程序Table 1. Program of gradient elution时间(min) 流动相A(%) 流动相B(%) 0.00 85.0 15.0 2.00 55.0 45.0 3.00 20.0 80.0 4.00 85.0 15.0 5.00 85.0 15.0 1.2.4 质谱条件
离子源为电喷雾离子源正离子模式(ESI+);毛细管电压3000 V;雾化器温度325 ℃;鞘气温度350 ℃;扫描模式为多反应监测(MRM),其他MRM参数见表2。
表 2 质谱参数Table 2. Mass spectrum parameter化合物 母离子(m/z) 子离子(m/z) 驻留时间(ms) 碎裂电压(V) 碰撞能(eV) 多西环素(DC) 445.2 428.1* 60 113 16 154.0 60 113 35 四环素(TC) 445.2 410.1* 60 150 20 427.1 60 150 10 土霉素(OTC) 461.2 426.1* 60 146 14 443.1 60 146 6 金霉素(CTC) 479.1 444.0* 60 141 12 462.0 60 141 20 四环素-D6
(TC-D6)451.0 416.0 60 150 20 注:带“*”为定量离子。 1.2.5 单因素实验
为了建立高效、准确的乌鳢肌肉中四环素类化合物残留分析方法,针对影响四种四环素类化合物回收率的因素,通过空白基质加标法,优化提取剂类型,分别采用10 mL甲醇、乙腈、酸化乙腈、酸化甲醇和酸化乙腈+2 mL Na2EDTA-McIlvanie缓冲液对加标的样品进行提取,比较其回收率;确定提取剂类型后对提取剂体积进行优化选择,分别采用5、10、15和20 mL提取剂提取乌鳢肌肉中目标化合物,以回收率为100%确定提取剂体积;分别采用100 mg的PSA、C18、GCB、中性氧化铝和弗罗里硅土对提取液进行净化,选择回收率较高且无杂质峰干扰的净化剂;对选择的净化剂用量进行优化选择,分别采用50、100、150、200、250和300 mg净化剂净化,比较其回收率和净化效果,每个处理设3次平行。
1.2.6 响应面优化
通过单因素实验获得对乌鳢肌肉中四环素类化合物回收率影响较大的因素进行响应面优化,以期获得最优的前处理方法。分别以PSA用量(X1)、C18用量(X2)和Na2EDTA-McIlvanie 缓冲液体积(X3)为自变量,四种四环素的各自回收率为应变量,采用Box-Behnken Design(BBD)法设计3因素3水平优化试验[26],试验设计因素与水平如表3所示。
表 3 Box-Behnken试验设计的编码水平和相应的自变量实际水平Table 3. Coded levels and corresponding actual levels of independent variables used for Box-Behnken design因素 水平 −1 0 1 X1 PSA用量(mg) 100 150 200 X2 C18用量(mg) 150 200 250 X3 Na2EDTA-McIlvanie 缓冲液体积(mL) 1 2 3 1.2.7 方法学验证
采用QuEChERS结合UHPLC-MS/MS法测定水产品中4种四环素类药物的残留量,用空白样品加标法评估方法检出限(LOD)和定量限(LOQ),在空白基质样品中添加4种化合物,分别以信噪比(S/N)大于或等于3和10的最低浓度为方法LOD和LOQ[27]。在空白基质中加入一定量的标准溶液后,采用建立的方法进行测定,通过加标样品的浓度与实际添加浓度的比值计算加标回收率,分别以LOQ、3倍LOQ和5倍LOQ为加标水平,每个水平重复6次,通过计算加标回收率和相对标准偏差验证方法的准确性和精密度;通过分析3 d内6个添加样品在5.0 μg/kg浓度下的回收率,计算相对标准偏差评价日间精密度。
采用提取后加标法,通过比较基质提取物和溶剂在0.5、1.0、2.0、5.0、10、20、50和100 ng/mL浓度下的校准曲线斜率评价基质效应(ME)。基质效应按照以下公式计算[28]:
ME(\%)= Sm−SsSs×100 (1) 式中,Sm为基质匹配标准溶液所得曲线的斜率,Ss为纯溶剂标准溶液所得曲线的斜率。当ME的值在−20%到+20%之间时,认为其基质效应是可接受的;如果它们介于−50%~−20%或+20%~+50%之间,则被认为是中等基质效应;如果这些值低于−50%或高于+50%则认为具有较强的基质效应[28]。
1.2.8 实际样品检测及风险评估
在广东乌鳢主要养殖区域的养殖池塘采集乌鳢样品,测定其肌肉中4种四环素类药物残留量。采用风险商(HQ,hazard quotient)对膳食中四环素类抗生素暴露量进行风险评估的表征。以人体重量为60 kg的每日允许受摄入量(ADI)为指标,对乌鳢中四环素类化合物的残留风险进行评估。为了评估膳食风险,根据Diao等[29]和Ramaswamy等[30]的方法计算了膳食暴露量(EDI)和风险商(HQ)值。通过将人均日消费量与公式(2)中所示的药物残留量相乘,计算出膳食暴露量(EDI)。通过计算公式(3)中所示的风险商(HQ)进行膳食风险评估[31−32]。
EDI = C×MBW (2) HQ = EDI/ADI (3) 式中,EDI为膳食暴露量(μg/(kg·bw·d)),C为样品中药物残留浓度(µg/kg),M为人均消费量(g),BW为成年平均体重60 kg;ADI 为每日允许摄入量(μg/(kg·bw·d))。当HQ值大于1时,风险被认为是不可接受的,而HQ值小于1则表示对人类的潜在风险较低[31]。
1.3 数据处理
实验数据采用Origin软件2021版本进行统计分析和图形输出,并使用Design-Expert软件12.0版本(State Ease, Inc.)进行响应面方差分析(ANOVA)和响应面图形输出,以确定线性项、二次项和交互项的回归系数,所有实验均进行3次重复。
2. 结果与分析
2.1 色谱条件的优化
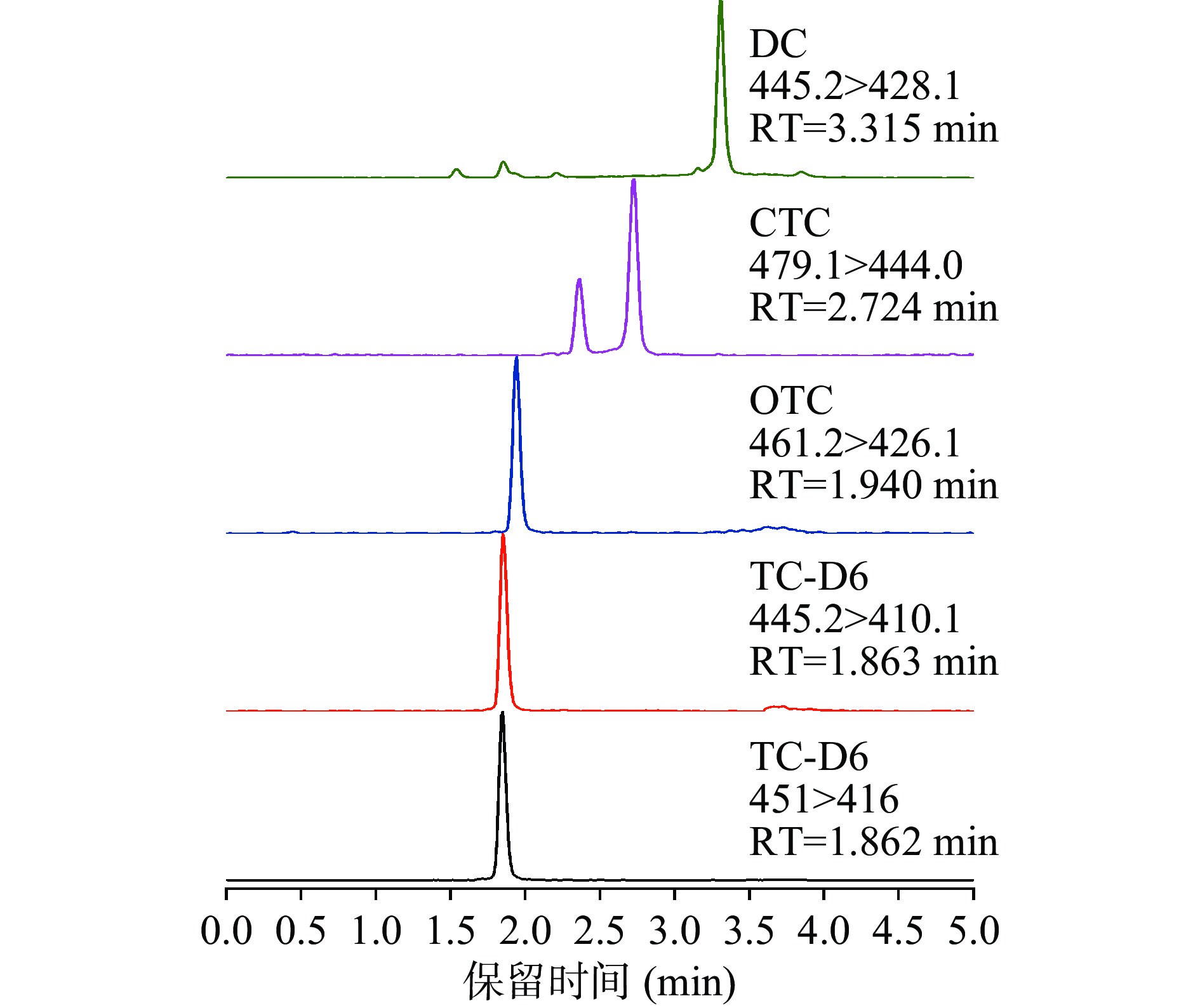
四环素类化合物分子中的双键和含氧或氮原子的基团可与其它物质相互作用[33],不仅可与金属离子形成二价、三价化合物,与蛋白质紧密结合,还可以与C18色谱柱上的硅醇基强烈作用,导致分离样品时出现拖尾峰[34]。因此需要选择合适的流动相,从而避免螯合物的形成及色谱柱的吸附,同时可以消除残留的金属离子[35−36],获得较好的分离度和理想的色谱峰。ESI+离子模式下在流动相中加入一定量的甲酸,可以增加目标物的离子化状态,提高其离子化效率,达到改善峰形,提高分离度的效果。为此对常用的酸化试剂甲酸、乙酸铵和乙酸进行了优化比较,结果表明在甲醇-0.5%甲酸水中四种四环素类化合物可获得较高的响应且峰形尖锐对称。同时对质谱的电离源、锥孔电压和碰撞能等参数进行优化,最终优化值见表2。通过对色谱条件的优化获得的四环素类化合物的色谱图峰形较好(图1)。
![]() 图 1 4种四环素类药物在加标浓度为5 μg/kg乌鳢肌肉样品中的MRM谱图Figure 1. MRM chromatograms of the 4 tetracycline drugs in muscle samples of Channa argus at 5 μg/kg supplemented concentration
图 1 4种四环素类药物在加标浓度为5 μg/kg乌鳢肌肉样品中的MRM谱图Figure 1. MRM chromatograms of the 4 tetracycline drugs in muscle samples of Channa argus at 5 μg/kg supplemented concentration2.2 前处理条件优化
2.2.1 提取溶剂选择
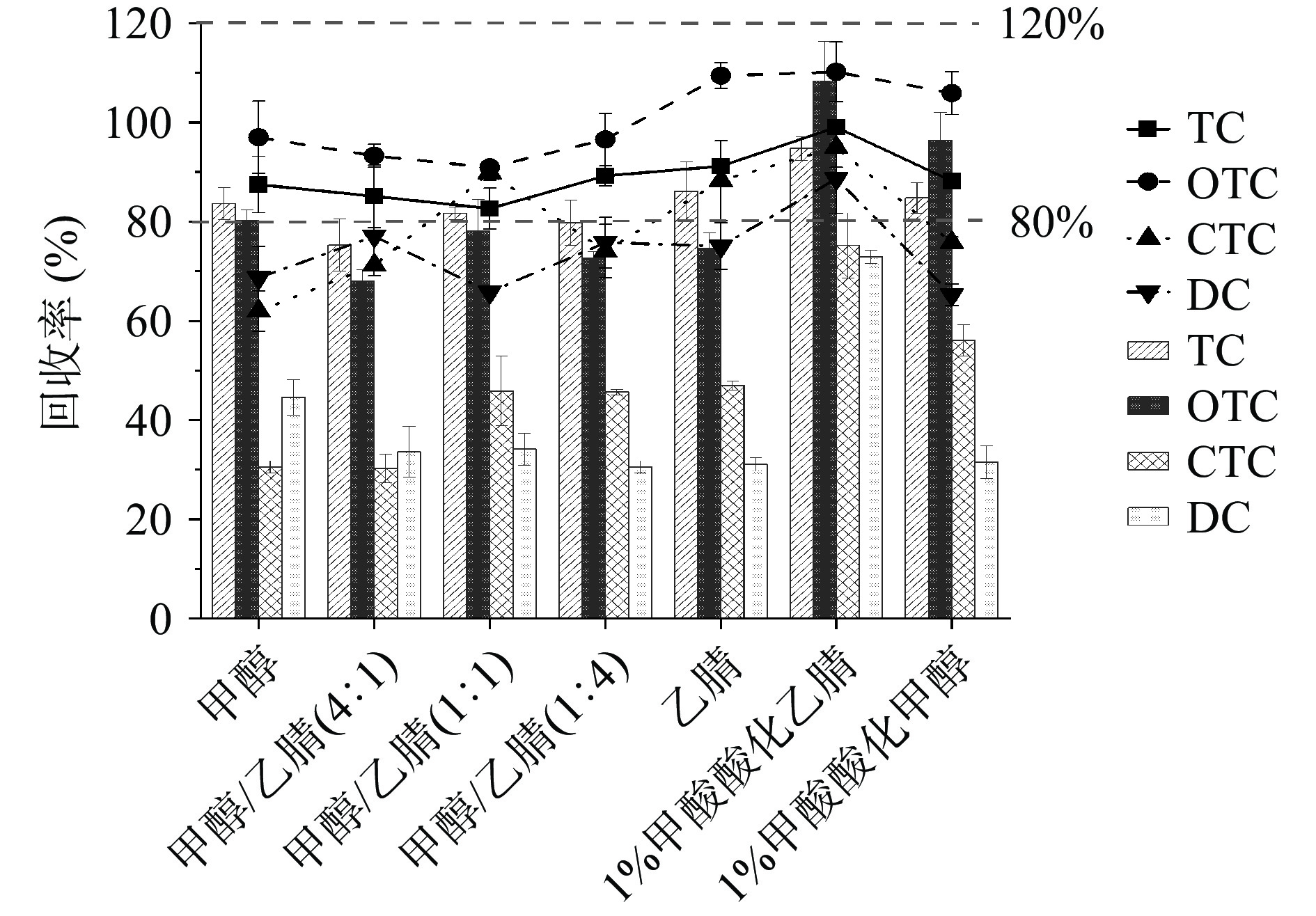
为了建立简单、通用和高效的样品前处理方法,获得较为洁净的提取物,对分析物具有较低的离子抑制,本研究对提取溶剂进行了优化选择。QuEChERS前处理方法常用的提取剂主要有甲醇、乙腈和酸化乙腈,因此采用加标回收法比较了甲醇、乙腈、酸化乙腈和酸化甲醇对水产品中四环素类化合物的提取效率(提取剂体积均为10 mL)。
不同提取剂对四种目标物的提取回收率见图2,结果表明,采用酸化乙腈和Na2EDTA-McIlvanie 缓冲液作为提取剂,四种四环素类化合物的提取效率较高,其加标回收率在80%~120%之间,可满足药物残留检测的要求。由于四环素类化合物可与基质中的二价金属离子(Ca2+、Mg2+、Zn2+和Cu2+)结合形成强配合物[37],需要在提取时阻止二价金属离子和蛋白质在提取过程中与四环素类化合物作用[8,38]。为了使目标物从金属配合物中释放出来,须在基质中加入EDTA或柠檬酸等螯合剂。在样品提取时加入Na2EDTA-McIlvanie(pH4.5)缓冲液作为金属离子螯合剂,可提高基质中四环素类化合物的提取效率。因此选择Na2EDTA-McIlvanie 缓冲液和1%甲酸乙腈作为四环素类药物的提取剂。
![]() 图 2 提取剂对四环素类化合物回收率的影响Figure 2. Effect of extraction agent on recovery of tetracyclines
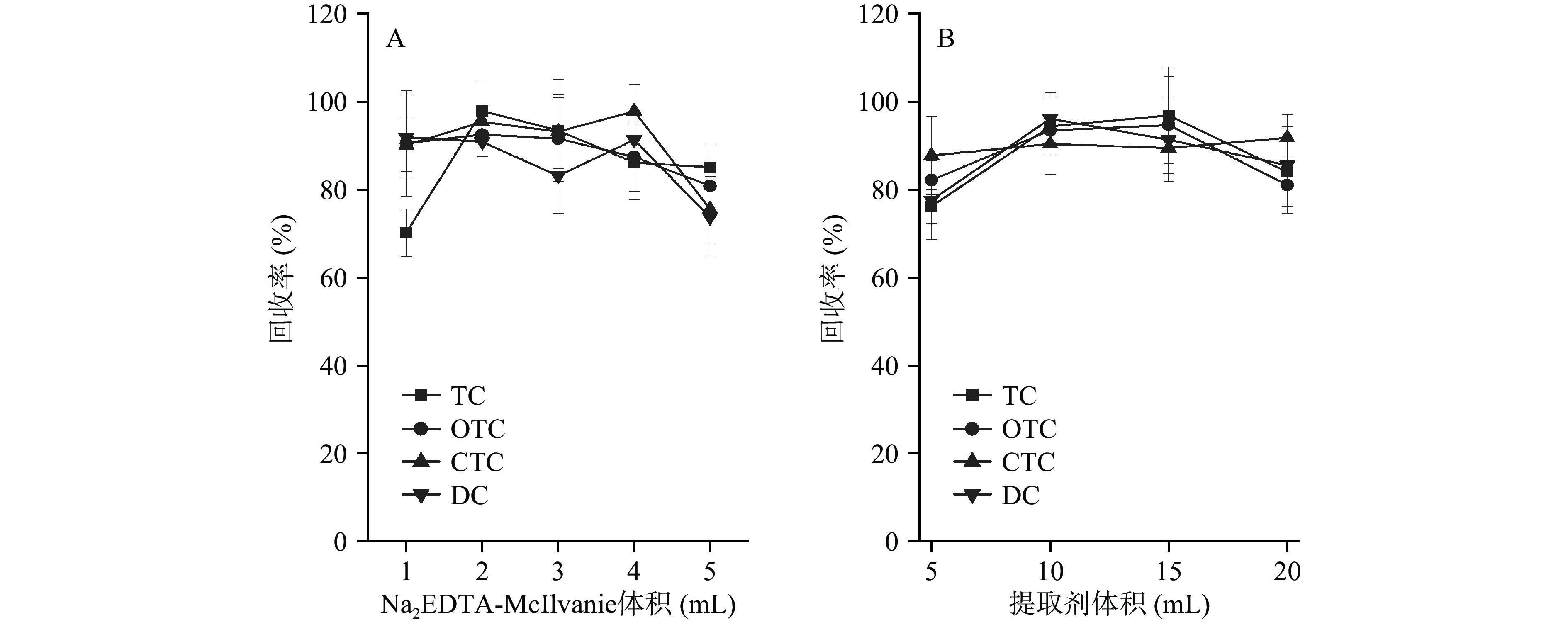
图 2 提取剂对四环素类化合物回收率的影响Figure 2. Effect of extraction agent on recovery of tetracyclines同时比较了Na2EDTA-McIlvanie 缓冲液体积分别为1、2、3、4和5 mL时四种化合物的回收率,结果如图3A所示,当缓冲液体积为2 mL 时可获得较高的回收率。此外比较了5、10、15 和20 mL提取剂对四种目标物回收率的影响(图3B),结果表明,5 mL 1%甲酸乙腈难以充分提取回收率较低,当酸化乙腈体积大于10 mL时回收率在80%~110%之间,因此选择1%甲酸乙腈的体积为10 mL。
![]() 图 3 Na2EDTA-McIlvanie和提取剂体积对四环素类化合物回收率的影响Figure 3. Effect of Na2EDTA-McIlvanie and extraction volume on recovery of tetracyclines
图 3 Na2EDTA-McIlvanie和提取剂体积对四环素类化合物回收率的影响Figure 3. Effect of Na2EDTA-McIlvanie and extraction volume on recovery of tetracyclines2.2.2 净化剂优化
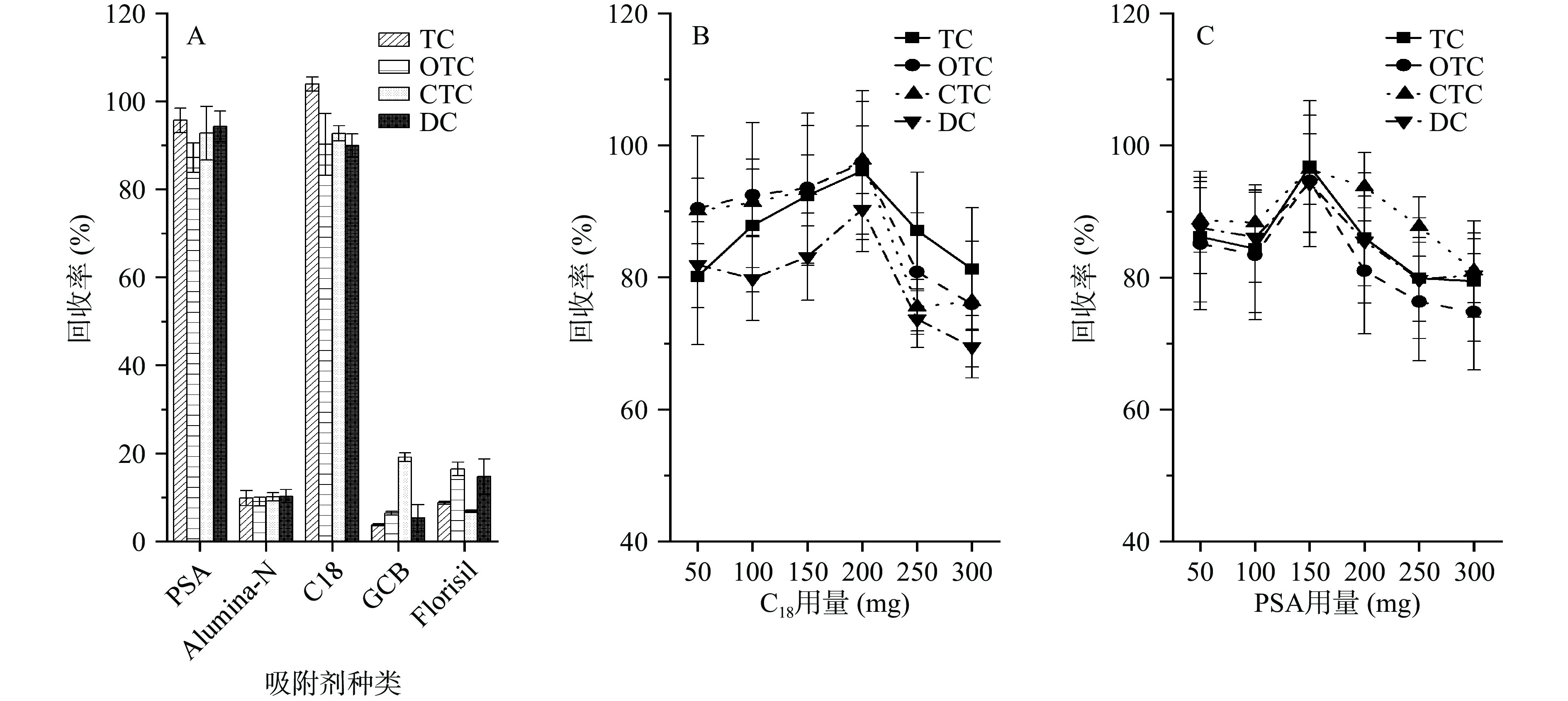
QuEChERS 方法常用的净化剂有PSA、C18、GCB、中性氧化铝(Alumina-N)和弗罗里硅土(Florisil),PSA通常用于去除提取液中的脂类和糖类物质,C18具有良好的除脂能力,GCB可去除提取液中的色素成分,但对含苯环官能团的化合物有较强的吸附作用。本研究优化比较了PSA、C18、GCB、中性氧化铝(Alumina-N)和弗罗里硅土(Florisil)5种吸附剂对4种四环素类药物的净化效果(图4A)。在空白乌鳢肌肉样品中加入同一浓度水平(5.0 µg/kg)的四环素类混合标准溶液,分别用100 mg的上述5种吸附剂净化,比较其回收率。结果表明,GCB、中性氧化铝(Alumina-N)和弗罗里硅土对四种四环素类抗菌药具有极强的吸附性能,三种吸附剂中四环素类药物的回收率低于20%,而在吸附剂PSA和C18净化下可获得较好回收率(80%~110%),因此选择PSA和C18作为前处理方法的吸附剂进行优化。
分别对吸附剂PSA和C18的用量进行优化比较,四环素类药物在不同剂量吸附剂下的回收率结果见图4B和图4C。结果表明,吸附剂用量对回收率有较大影响。从图4B可知,吸附剂C18的用量对四种四环素类药物的回收率有较大影响,C18用量从50 mg增加至200 mg时回收率逐渐增加,当C18用量为200 mg时回收率较为理想(90.3%~97.8%),但C18用量大于250 mg时,回收率明显降低。PSA用量同样也影响回收率,当PSA用量为150 mg时,四种药物回收率在94.3%~96.8%之间,但是PSA用量在100 mg以下或200 mg以上时回收率偏低。因此选择C18用量分别为150、200和250 mg,PSA用量为100、150和200 mg进行响应面优化。
![]() 图 4 吸附剂种类和用量对加标回收率的影响Figure 4. The influence of type and dosage of cleaning agents on recoveries
图 4 吸附剂种类和用量对加标回收率的影响Figure 4. The influence of type and dosage of cleaning agents on recoveries2.3 响应面优化
在单因素实验结果基础上,选择PSA用量(X1)、C18用量(X2)和Na2EDTA-McIlvanie缓冲液体积(X3)进行三因素三水平的Box-Benken响应面优化试验(BBD),响应值为四环素(Y1)、土霉素(Y2)、金霉素(Y3)和多西环素(Y4)的回收率,结果见表4。
表 4 Box-Benken响应面优化试验方案与结果Table 4. Experimental scheme and results of Box-Behnken design试验号 X1 X2 X3 Y1(%) Y2(%) Y3(%) Y4(%) 1 0 0 0 102.6 103.4 98.7 98.8 2 1 −1 0 81.6 87.8 80.6 80.8 3 0 0 0 106.9 104.9 101.1 96.9 4 1 1 0 77.7 87.9 70.3 71.3 5 −1 1 0 86.1 86.6 70.0 80.9 6 0 0 0 97.6 103.2 103.7 99.2 7 −1 0 1 82.0 79.7 68.1 72.65 8 −1 0 −1 90.9 84.6 70.8 78.8 9 0 0 0 96.8 102.4 95.5 105.2 10 −1 −1 0 86.3 84.7 83.5 84.3 11 1 0 1 81.4 89.1 75.0 78.7 12 0 −1 −1 78.5 86.1 86.9 84.3 13 0 −1 1 76.5 82.7 82.0 77.4 14 0 1 1 77.4 85.0 67.1 75.5 15 1 1 −1 74.0 84.1 70.4 73.3 16 0 0 0 98.3 99.8 92.4 96.0 17 0 1 −1 73.4 86.1 76.9 75.4 2.3.1 模型建立及方差分析
对BBD实验设计的结果进行了模型拟合和统计分析,以预测最佳QuEChERS条件。采用Design-Expert 12.0软件将实验数据拟合成二阶多项式模型,通过多元回归分析各自变量与响应之间的关系,获得4种四环素类药物的拟合回归方程:
四环素(TC):Y1=−146.72+0.55X1+1.82X2+31.54X3−0.0004X1X2+0.082X1X3+0.030X2X3−0.0024X12−0.0046X22−12.42X32
土霉素(OTC):Y2=−127.14+0.966X1+1.24X2+29.99X3−0.0019X1X2+0.0498X1X3+0.0121X2X3−0.0033X12−0.0031X22−10.08X32
金霉素(CTC):Y3=−166.93+1.63X1+1.08X2+47.98X3+0.0003X1X2+0.037X1X3−0.0246X2X3−0.0059X12−0.0030X22−12.53X32
多西环素(DC):Y4=−158.37+1.30X1+1.37X2+32.36X3−0.0006X1X2+0.0585X1X3+0.0355X2X3−0.0044X12−0.0035X22−12.30X32
为了检验模型的拟合程度,对模型进行方差分析和显著性检验,方差分析由Fisher F检验进行推导[39],结果见表5。一般而言F值越高,P值越低,回归模型越显著[40],四种药物回归模型F值在14.59~46.43之间(自由度df=9),P值均远低于0.01,回归模型差异极显著(表5)。四种药物回归方程的回归系数R2分别为0.9574(TC)、0.9835(OTC)、0.9494(CTC)和0.9575(DC),调整回归系数R2adj分别为0.9027(TC)、0.9623(OTC)、0.8843(CTC)和0.9028(DC),表明模型拟合良好。且四种药物模型失拟值不显著,F值较低(0.0738~0.7510),P值均大于0.05(0.5762~0.9709),表明回归模型在本研究中可行[39],具有较好的准确性和可靠性,可用来分析和预测各因素对4种目标物回收率的影响。
表 5 四环素类药物回收率响应面模型及预测结果方差分析Table 5. ANOVA of response surface model and predicted results for recovery of tetracyclines来源 四环素(TC) 土霉素(OTC) 金霉素(CTC) 多西环素(DC) F P F P F P F P 模型 17.49 0.0005** 46.43 <0.0001** 14.59 0.0009** 17.51 0.0005** X1 10.82 0.0133* 8.19 0.0243* 0.11 0.7527 1.69 0.2350 X2 0.7995 0.4010 0.8544 0.3861 16.66 0.0047** 6.17 0.0419* X3 0.0028 0.9595 0.9068 0.3727 1.15 0.3195 0.63 0.4531 X1X2 0.3104 0.5948 0.3197 0.5894 0.14 0.7148 0.80 0.4009 X1X3 6.13 0.0425* 8.93 0.0203* 0.75 0.4155 2.99 0.1273 X2X3 0.8185 0.3957 0.5412 0.4858 0.34 0.5780 1.10 0.3288 X12 13.70 0.0076** 106.79 <0.0001** 50.82 0.0002** 45.26 0.0003** X22 51.85 0.0002** 91.94 <0.0001** 13.32 0.0082** 28.43 0.0011** X32 59.85 0.0001** 158.25 <0.0001** 37.19 0.0005** 55.65 0.0001** 失拟值 0.0738 0.9709 0.4760 0.7159 0.7510 0.5762 0.7432 0.5797 R2adj/R2 0.9027 0.9574 0.9623 0.9835 0.8843 0.9494 0.9028 0.9575 预测值(%) 100.0 102 99.4 99.4 实验值(%) 101±1.68 97.7±3.82 101±3.06 95.7±2.75 注:“**”表示差异极显著(P<0.01),“*”表示差异显著(P<0.05)。 2.3.2 响应面分析
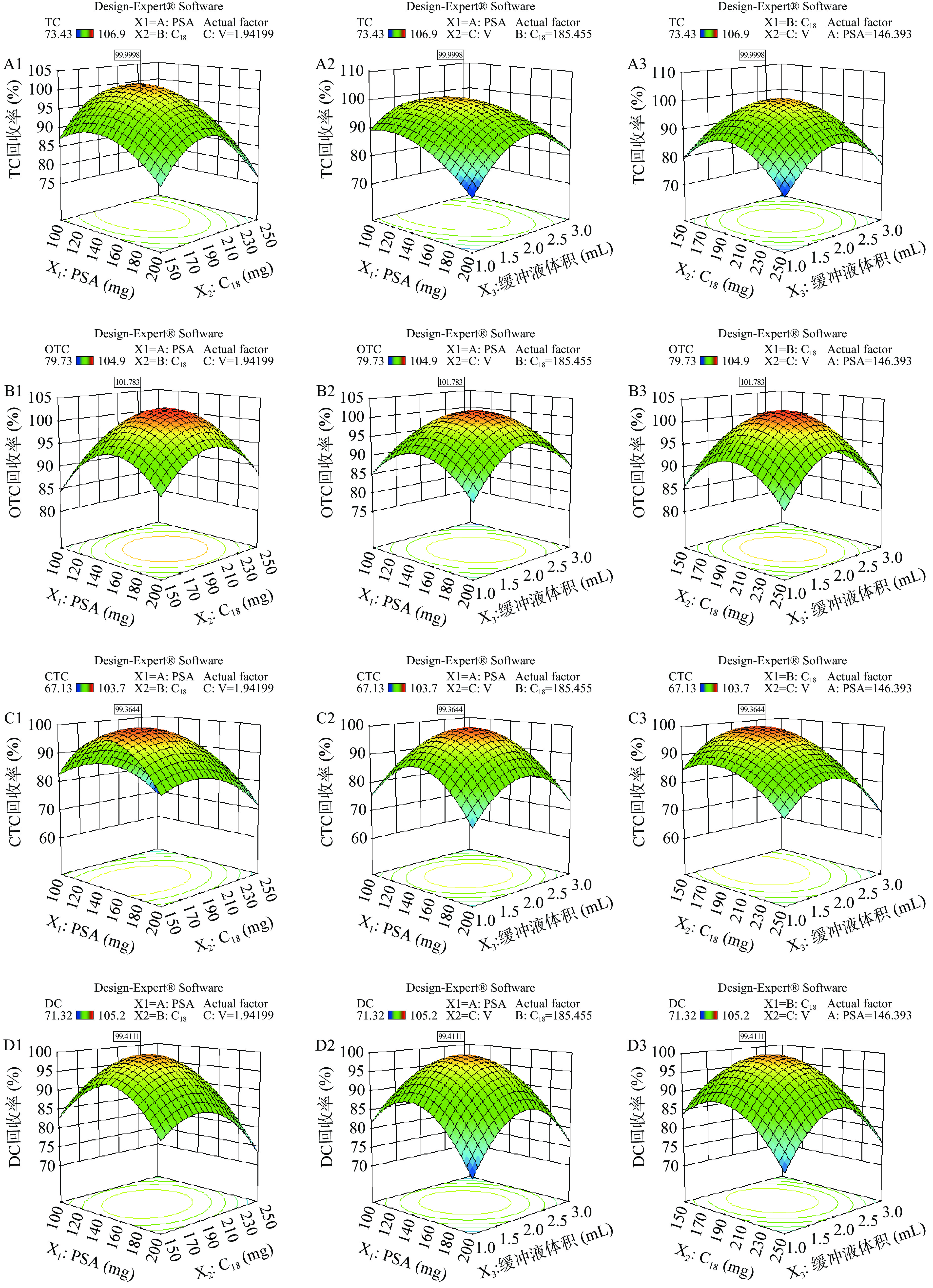
为了最大限度的提高方法回收率,根据拟合方程进行响应面分析,绘制各变量之间的交互作用三维响应面曲线,四种四环素类药物的回收率受PSA用量(X1)、C18用量(X2)和Na2EDTA-McIlvanie 缓冲液体积(X3)的影响如图5(TC:A1~A3,OTC:B1~B3,CTC:C1~C3,DC:D1~D3)所示。响应面的斜率越大,表明该因素对响应的影响更大[41];响应面曲面可直观反映各因素间的交互作用对目标物回收率的影响,图中因素的曲面越陡表明该因素对目标物回收率影响越大。如由图5可知,对于四环素和土霉素PSA用量(X1)和Na2EDTA-McIlvanie 缓冲液体积(X3)的响应面曲面比较陡,表明两者间交互作用显著;而PSA用量(X2)和C18用量(X2)的响应面曲线比较缓,表明两者之间交互作用不明显。
![]() 图 5 各因素交互作用对回收率影响的响应面图注:A:四环素、B:土霉素、C:金霉素、D:多西环素。Figure 5. Response surface estimated showing the interaction of two different parameters on the recovery
图 5 各因素交互作用对回收率影响的响应面图注:A:四环素、B:土霉素、C:金霉素、D:多西环素。Figure 5. Response surface estimated showing the interaction of two different parameters on the recovery通过Design-Expert 12.0软件分析,以四环素类化合物的回收率均为100%,确定最佳QuEChERS 条件为PSA:146.4 mg、C18:185.5 mg和Na2EDTA-McIlvanie 缓冲液体积:1.95 mL,该最优条件下的四环素、土霉素、金霉素和多西环素的回收率预测值分别为:100%、102%、99.4%、99.4%。调整实际条件最佳QuEChERS 条件为PSA:150 mg、C18:200 mg和Na2EDTA-McIlvanie 缓冲液体积:2.0 mL,采用优化后的参数进行验证实验,经6次平行实验后测得平均回收率分别为:101±1.7%(TC)、97.7±3.8%(OTC)、101±3.1%(CTC)和95.7±2.8%(DC),四种化合物实际回收率与模型预测值差异不明显,说明拟合模型优化出的QuEChERS条件较为准确。
2.4 方法学验证
2.4.1 方法的线性、检出限、定量限
在上述优化的仪器条件下,分析一系列不同浓度的基质样品,以各化合物的质量浓度为横坐标,定量离子峰面积为纵坐标,绘制标准曲线。结果表明,四种四环素类药物在线性范围内均呈良好的线性关系(r>0.999)。添加不同浓度混合标准溶液至空白样品进行测试,分别以目标峰S/N=3和10确定检出限(LOD)和定量限(LOQ),结果如表6所示。在0.5~100 ng/mL 浓度范围内,4种四环素类化合物的线性关系良好,相关系数均大于0.999;检出限为0.25~0.75 μg/kg,定量限为0.84~2.00 μg/kg,满足国内外药物残留检测要求。
表 6 四环素类化合物线性方程、相关系数、检出限、定量限和基质效应Table 6. Linear equation, correlation coefficient, LOD, LOQ and matrix effects for tetracyclines化合物 线性范围(ng/mL) 基质 线性方程 相关系数(r) LOD(µg/kg) LOQ(µg/kg) 基质效应(%) 四环素(TC) 0.5~100 溶剂 Y=0.0453X−0.0087 0.9995 0.50 1.50 − 乌鳢 Y=0.0477X−0.0176 0.9998 0.75 2.00 5.30 土霉素(OTC) 0.5~100 溶剂 Y=0.0245X−0.0144 0.9997 0.46 1.50 − 乌鳢 Y=0.0186X−0.0032 0.9997 0.50 1.50 −24.1 金霉素(CTC) 0.5~100 溶剂 Y=0.0195X−0.0113 0.9995 0.25 0.89 − 乌鳢 Y=0.0179X−0.0058 0.9991 0.30 1.00 −8.21 多西素(DC) 0.5~100 溶剂 Y=0.0386X−0.0112 0.9999 0.25 0.84 − 乌鳢 Y=0.0247X+0.0637 0.9988 0.30 1.00 −36.0 2.4.2 回收率和精密度
采用基质加标法对方法回收率和精密度进行了考察,分设置LOQ、3倍LOQ和5倍LOQ三种添加浓度水平,按照本方法确定的前处理方法和仪器条件进行测定,每个浓度水平设置6次重复,结果见表7。四种四环素类化合物日内和日间回收率在89.5%~118%之间,精密度小于10%,与现有文献报道的方法相比(表8),本方法回收率和精密度较好,可满足国内外药物残留检测要求。
表 7 四环素类抗生素在乌鳢肌肉中的加标回收率和精密度(n=6)Table 7. Recovery and precision of tetracyclines in the muscle of Channa argus (n=6)化合物 添加水平(μg/kg) 日内 日间 平均回收率(%) RSD(%) 平均回收率(%) RSD(%) 土霉素(OTC) 1.5 91.5 6.28 105 5.95 4.5 101 1.43 91.0 7.28 7.5 94.3 4.77 95.3 3.33 四环素(TC) 2.0 89.5 3.63 90.1 8.68 6.0 96.4 3.31 92.1 8.59 10 94.8 1.11 98.1 3.15 金霉素(CTC) 1.0 105 2.90 103 2.79 3.0 108 3.27 91.7 6.69 5.0 108 6.34 94.3 4.30 多西环素(DC) 1.0 95.5 2.31 107 2.86 3.0 111 8.88 96.1 2.25 5.0 109 4.20 118 4.52 表 8 本方法与其他报道的水产品中四环素类药物残留检测方法比较Table 8. Comparison of the proposed method with other reported methods for the determination of TCs in fish samples2.4.3 基质效应
采用空白基质提取后加标法,用空白乌鳢肌肉样品按照优化的前处理方法进行前处理后,分别采用定溶液(15%甲醇水)和空白基质溶液配制0.5~100 ng/mL的标准溶液,按照优化好的仪器条件进行测定,计算乌鳢基质中四环素类化合物的基质效应,结果见表6。四种四环素类化合物的基质效应在−36.0%~5.30%之间,其中四环素存在基质增强效应,其他三种物质存在基质抑制效应,为减少基质效应对检测结果的影响,在实际样品检测中应采用基质标准曲线进行定量校准。
2.5 实际样品测定及风险分析
从广东省乌鳢主要养殖区域采集乌鳢肌肉样品33个,按照本研究优化的方法检测样品中四环素类化合物的残留量,并通过计算风险商(hazard quotient,HQ)对4种四环素类化合物的残留风险进行风险评估。乌鳢样品中四环素类化合物残留结果见表9。
表 9 乌鳢肌肉中四环素类抗生素残留浓度和风险商Table 9. The residual concentrations and HQ of tetracyclines in the muscle of Channa argus样品编号 土霉素(OTC) 四环素(TC) 金霉素(CTC) 多西环素(DC) 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ S001 5.04 1.07×10−4 4.69 9.92×10−5 N.D. − N.D. − S002 5.24 1.11×10−4 4.02 8.50×10−5 16.86 3.57×10−4 N.D. − S003 N.D. − 4.17 8.82×10−5 N.D. − N.D. − S004 N.D. − 3.7 7.83×10−5 N.D. − N.D. − S005 N.D. − 4.42 9.35×10−5 N.D. − N.D. − S006 N.D. − 4.28 9.05×10−5 N.D. − N.D. − S007 N.D. − 3.71 7.85×10−5 N.D. − N.D. − S008 N.D. − 4.07 8.61×10−5 N.D. − N.D. − S009 N.D. − N.D. N.D. N.D. − N.D. − S010 N.D. − 4.44 9.39×10−5 N.D. − 14.52 3.07×10−3 S011 N.D. − 3.64 7.70×10−5 7.1 1.50×10−4 2.66 5.63×10−4 S012 N.D. − 3.69 7.81×10−5 N.D. − 1.47 3.11×10−4 S013 N.D. − 6.43 1.36×10−4 N.D. − 1.25 2.64×10−4 S014 N.D. − 3.57 7.55×10−5 N.D. − N.D. − S015 N.D. − 3.69 7.81×10−5 N.D. − N.D. − S016 N.D. − N.D. N.D. N.D. − N.D. − S017 7.52 1.59×10−4 3.74 7.91×10−5 N.D. − N.D. − S018 N.D. − 3.55 7.51×10−5 N.D. − N.D. − S019 N.D. − 3.8 8.04×10−5 N.D. − N.D. − S020 N.D. − 4.17 8.82×10−5 N.D. − 1.08 2.28×10−4 S021 N.D. − 3.72 7.87×10−5 N.D. − 2.55 5.39×10−4 S022 5.17 1.09×10−4 4.22 8.93×10−5 N.D. − N.D. − S023 8.4 1.78×10−4 5.81 1.23×10−4 2.52 5.33×10−5 N.D. − S024 N.D. − N.D. N.D. N.D. − N.D. − S025 N.D. − 3.55 7.51×10−5 N.D. − N.D. − S026 N.D. − 3.57 7.55×10−5 N.D. − 6.46 1.37×10−3 S027 N.D. − 3.7 7.83×10−5 N.D. − N.D. − S028 N.D. − 3.57 7.55×10−5 N.D. − N.D. − S029 N.D. − 5.48 1.16×10−4 34.56 7.31×10−4 3.34 7.07×10−4 S030 N.D. − 3.69 7.81×10−5 N.D. − N.D. − S031 N.D. − 4.45 9.41×10−5 N.D. − N.D. − S032 N.D. − 3.73 7.89×10−5 N.D. − N.D. − S033 N.D. − 4.88 9.92×10−5 N.D. − N.D. − 注:N.D.表示未检出;−表示没有值。 以我国制定的抗生素每日允许摄入量(Acceptable Daily Intake,ADI μg/(kg·bw·d))为标准进行评价,四环素、金霉素和土霉素ADI均为30 μg/(kg·bw·d),多西环素ADI为3.0 μg/(kg·bw·d)。根据国家统计局数据,2020年我国人均水产品消费量为38.08 g/d,通过计算可知,土霉素HQ值在1.07×10−4~1.78×10−4之间,四环素HQ值在7.51×10−5~1.36×10−4,金霉素HQ值在5.33×10−5~7.31×10−4,多西环素HQ值为2.28×10−4~3.07×10−3。所有四环素类化合物HQ值均远低于1.0,表明通过食用广东养殖乌鳢摄入四环素类药物对人体的健康没有影响,风险较低,膳食安全性较高。
3. 结论
本研究采用响应面法优化了四环素类抗生素在乌鳢肌肉中的QuEChERS结合超高效液相色谱-串联质谱(UHPLC-MS/MS)检测方法,样品用10 mL 1%甲酸酸化乙腈和2 mL 0.1 mol/L Na2EDTA-McIlvaine(pH4.5)缓冲溶液提取,经150 mg PSA和200 mg C18 净化后,采用UHPLC-MS/MS测定,内标法定量。结果表明,该方法简便快捷、溶剂用量少,且准确性和精密度较高。在0.5~100 ng/mL浓度范围内线性良好,相关系数均大于0.999;检出限为0.25~0.75 μg/kg,定量限为0.84~2.00 μg/kg。在三个添加浓度水平下四种四环素类化合物的日内和日间回收率在89.5%~118%之间,精密度小于10%,满足国内外药物残留检测要求,可用于乌鳢肌肉中四环素类药物残留检测。本方法的定量限较已有报道的方法低,回收率、精密度和基质效应与现有方法基本一致。采用优化的快速检测方法检测了广东省乌鳢主要养殖区域的33个乌鳢样品,通过计算风险商(HQ)进行风险评估,结果表明广东乌鳢中四环素的膳食风险较低,其残留对人体健康无影响。
-
![]()
图 1 4种四环素类药物在加标浓度为5 μg/kg乌鳢肌肉样品中的MRM谱图
Figure 1. MRM chromatograms of the 4 tetracycline drugs in muscle samples of Channa argus at 5 μg/kg supplemented concentration
![]()
图 2 提取剂对四环素类化合物回收率的影响
Figure 2. Effect of extraction agent on recovery of tetracyclines
![]()
图 3 Na2EDTA-McIlvanie和提取剂体积对四环素类化合物回收率的影响
Figure 3. Effect of Na2EDTA-McIlvanie and extraction volume on recovery of tetracyclines
![]()
图 4 吸附剂种类和用量对加标回收率的影响
Figure 4. The influence of type and dosage of cleaning agents on recoveries
![]()
图 5 各因素交互作用对回收率影响的响应面图
注:A:四环素、B:土霉素、C:金霉素、D:多西环素。
Figure 5. Response surface estimated showing the interaction of two different parameters on the recovery
表 1 梯度洗脱程序
Table 1 Program of gradient elution
时间(min) 流动相A(%) 流动相B(%) 0.00 85.0 15.0 2.00 55.0 45.0 3.00 20.0 80.0 4.00 85.0 15.0 5.00 85.0 15.0  下载: 导出CSV
下载: 导出CSV
表 2 质谱参数
Table 2 Mass spectrum parameter
化合物 母离子(m/z) 子离子(m/z) 驻留时间(ms) 碎裂电压(V) 碰撞能(eV) 多西环素(DC) 445.2 428.1* 60 113 16 154.0 60 113 35 四环素(TC) 445.2 410.1* 60 150 20 427.1 60 150 10 土霉素(OTC) 461.2 426.1* 60 146 14 443.1 60 146 6 金霉素(CTC) 479.1 444.0* 60 141 12 462.0 60 141 20 四环素-D6
(TC-D6)451.0 416.0 60 150 20 注:带“*”为定量离子。
下载: 导出CSV
表 3 Box-Behnken试验设计的编码水平和相应的自变量实际水平
Table 3 Coded levels and corresponding actual levels of independent variables used for Box-Behnken design
因素 水平 −1 0 1 X1 PSA用量(mg) 100 150 200 X2 C18用量(mg) 150 200 250 X3 Na2EDTA-McIlvanie 缓冲液体积(mL) 1 2 3
下载: 导出CSV
表 4 Box-Benken响应面优化试验方案与结果
Table 4 Experimental scheme and results of Box-Behnken design
试验号 X1 X2 X3 Y1(%) Y2(%) Y3(%) Y4(%) 1 0 0 0 102.6 103.4 98.7 98.8 2 1 −1 0 81.6 87.8 80.6 80.8 3 0 0 0 106.9 104.9 101.1 96.9 4 1 1 0 77.7 87.9 70.3 71.3 5 −1 1 0 86.1 86.6 70.0 80.9 6 0 0 0 97.6 103.2 103.7 99.2 7 −1 0 1 82.0 79.7 68.1 72.65 8 −1 0 −1 90.9 84.6 70.8 78.8 9 0 0 0 96.8 102.4 95.5 105.2 10 −1 −1 0 86.3 84.7 83.5 84.3 11 1 0 1 81.4 89.1 75.0 78.7 12 0 −1 −1 78.5 86.1 86.9 84.3 13 0 −1 1 76.5 82.7 82.0 77.4 14 0 1 1 77.4 85.0 67.1 75.5 15 1 1 −1 74.0 84.1 70.4 73.3 16 0 0 0 98.3 99.8 92.4 96.0 17 0 1 −1 73.4 86.1 76.9 75.4
下载: 导出CSV
表 5 四环素类药物回收率响应面模型及预测结果方差分析
Table 5 ANOVA of response surface model and predicted results for recovery of tetracyclines
来源 四环素(TC) 土霉素(OTC) 金霉素(CTC) 多西环素(DC) F P F P F P F P 模型 17.49 0.0005** 46.43 <0.0001** 14.59 0.0009** 17.51 0.0005** X1 10.82 0.0133* 8.19 0.0243* 0.11 0.7527 1.69 0.2350 X2 0.7995 0.4010 0.8544 0.3861 16.66 0.0047** 6.17 0.0419* X3 0.0028 0.9595 0.9068 0.3727 1.15 0.3195 0.63 0.4531 X1X2 0.3104 0.5948 0.3197 0.5894 0.14 0.7148 0.80 0.4009 X1X3 6.13 0.0425* 8.93 0.0203* 0.75 0.4155 2.99 0.1273 X2X3 0.8185 0.3957 0.5412 0.4858 0.34 0.5780 1.10 0.3288 X12 13.70 0.0076** 106.79 <0.0001** 50.82 0.0002** 45.26 0.0003** X22 51.85 0.0002** 91.94 <0.0001** 13.32 0.0082** 28.43 0.0011** X32 59.85 0.0001** 158.25 <0.0001** 37.19 0.0005** 55.65 0.0001** 失拟值 0.0738 0.9709 0.4760 0.7159 0.7510 0.5762 0.7432 0.5797 R2adj/R2 0.9027 0.9574 0.9623 0.9835 0.8843 0.9494 0.9028 0.9575 预测值(%) 100.0 102 99.4 99.4 实验值(%) 101±1.68 97.7±3.82 101±3.06 95.7±2.75 注:“**”表示差异极显著(P<0.01),“*”表示差异显著(P<0.05)。
下载: 导出CSV
表 6 四环素类化合物线性方程、相关系数、检出限、定量限和基质效应
Table 6 Linear equation, correlation coefficient, LOD, LOQ and matrix effects for tetracyclines
化合物 线性范围(ng/mL) 基质 线性方程 相关系数(r) LOD(µg/kg) LOQ(µg/kg) 基质效应(%) 四环素(TC) 0.5~100 溶剂 Y=0.0453X−0.0087 0.9995 0.50 1.50 − 乌鳢 Y=0.0477X−0.0176 0.9998 0.75 2.00 5.30 土霉素(OTC) 0.5~100 溶剂 Y=0.0245X−0.0144 0.9997 0.46 1.50 − 乌鳢 Y=0.0186X−0.0032 0.9997 0.50 1.50 −24.1 金霉素(CTC) 0.5~100 溶剂 Y=0.0195X−0.0113 0.9995 0.25 0.89 − 乌鳢 Y=0.0179X−0.0058 0.9991 0.30 1.00 −8.21 多西素(DC) 0.5~100 溶剂 Y=0.0386X−0.0112 0.9999 0.25 0.84 − 乌鳢 Y=0.0247X+0.0637 0.9988 0.30 1.00 −36.0
下载: 导出CSV
表 7 四环素类抗生素在乌鳢肌肉中的加标回收率和精密度(n=6)
Table 7 Recovery and precision of tetracyclines in the muscle of Channa argus (n=6)
化合物 添加水平(μg/kg) 日内 日间 平均回收率(%) RSD(%) 平均回收率(%) RSD(%) 土霉素(OTC) 1.5 91.5 6.28 105 5.95 4.5 101 1.43 91.0 7.28 7.5 94.3 4.77 95.3 3.33 四环素(TC) 2.0 89.5 3.63 90.1 8.68 6.0 96.4 3.31 92.1 8.59 10 94.8 1.11 98.1 3.15 金霉素(CTC) 1.0 105 2.90 103 2.79 3.0 108 3.27 91.7 6.69 5.0 108 6.34 94.3 4.30 多西环素(DC) 1.0 95.5 2.31 107 2.86 3.0 111 8.88 96.1 2.25 5.0 109 4.20 118 4.52
下载: 导出CSV
表 8 本方法与其他报道的水产品中四环素类药物残留检测方法比较
Table 8 Comparison of the proposed method with other reported methods for the determination of TCs in fish samples
下载: 导出CSV
表 9 乌鳢肌肉中四环素类抗生素残留浓度和风险商
Table 9 The residual concentrations and HQ of tetracyclines in the muscle of Channa argus
样品编号 土霉素(OTC) 四环素(TC) 金霉素(CTC) 多西环素(DC) 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ 残留浓度(μg/kg) HQ S001 5.04 1.07×10−4 4.69 9.92×10−5 N.D. − N.D. − S002 5.24 1.11×10−4 4.02 8.50×10−5 16.86 3.57×10−4 N.D. − S003 N.D. − 4.17 8.82×10−5 N.D. − N.D. − S004 N.D. − 3.7 7.83×10−5 N.D. − N.D. − S005 N.D. − 4.42 9.35×10−5 N.D. − N.D. − S006 N.D. − 4.28 9.05×10−5 N.D. − N.D. − S007 N.D. − 3.71 7.85×10−5 N.D. − N.D. − S008 N.D. − 4.07 8.61×10−5 N.D. − N.D. − S009 N.D. − N.D. N.D. N.D. − N.D. − S010 N.D. − 4.44 9.39×10−5 N.D. − 14.52 3.07×10−3 S011 N.D. − 3.64 7.70×10−5 7.1 1.50×10−4 2.66 5.63×10−4 S012 N.D. − 3.69 7.81×10−5 N.D. − 1.47 3.11×10−4 S013 N.D. − 6.43 1.36×10−4 N.D. − 1.25 2.64×10−4 S014 N.D. − 3.57 7.55×10−5 N.D. − N.D. − S015 N.D. − 3.69 7.81×10−5 N.D. − N.D. − S016 N.D. − N.D. N.D. N.D. − N.D. − S017 7.52 1.59×10−4 3.74 7.91×10−5 N.D. − N.D. − S018 N.D. − 3.55 7.51×10−5 N.D. − N.D. − S019 N.D. − 3.8 8.04×10−5 N.D. − N.D. − S020 N.D. − 4.17 8.82×10−5 N.D. − 1.08 2.28×10−4 S021 N.D. − 3.72 7.87×10−5 N.D. − 2.55 5.39×10−4 S022 5.17 1.09×10−4 4.22 8.93×10−5 N.D. − N.D. − S023 8.4 1.78×10−4 5.81 1.23×10−4 2.52 5.33×10−5 N.D. − S024 N.D. − N.D. N.D. N.D. − N.D. − S025 N.D. − 3.55 7.51×10−5 N.D. − N.D. − S026 N.D. − 3.57 7.55×10−5 N.D. − 6.46 1.37×10−3 S027 N.D. − 3.7 7.83×10−5 N.D. − N.D. − S028 N.D. − 3.57 7.55×10−5 N.D. − N.D. − S029 N.D. − 5.48 1.16×10−4 34.56 7.31×10−4 3.34 7.07×10−4 S030 N.D. − 3.69 7.81×10−5 N.D. − N.D. − S031 N.D. − 4.45 9.41×10−5 N.D. − N.D. − S032 N.D. − 3.73 7.89×10−5 N.D. − N.D. − S033 N.D. − 4.88 9.92×10−5 N.D. − N.D. − 注:N.D.表示未检出;−表示没有值。
下载: 导出CSV
-
[1] 李俊锁, 邱月明, 王超. 兽药残留分析[M]. 上海:上海科学技术出版社, 2002:365−390 LI J S, QIU Y M, WANG C. Analysis of veterinary drug residues [M]. Shanghai:Shanghai Scientific & Technical Publishers, 2002:365−390.
[2] LI S, SHI WZ, LIU W, et al. A duodecennial national synthesis of antibiotics in China's major rivers and seas (2005-2016)[J]. Sci Total Environ,2018,615:906−917. doi: 10.1016/j.scitotenv.2017.09.328
[3] ZHANG M, LIU Y S, ZHAO J L, et al. Occurrence, fate and mass loadings of antibiotics in two swine wastewater treatment systems[J]. Sci Total Environ,2018,639:1421−1431. doi: 10.1016/j.scitotenv.2018.05.230
[4] 罗方园, 潘根兴, 李恋卿, 等. 洪泽湖沉积物中四环素土霉素及相关抗性基因的分布特征及潜在风险分析[J]. 农业环境科学学报,2017,36(2):369−375 doi: 10.11654/jaes.2016-1237 LUO F Y, PAN G X, LI L Q, et al. The distribution characteristics and potential risk of tetracycline, oxytetracycline, and their corresponding gene pollution in sediment of Hongze Lake[J]. Journal of Agro-Environment Science,2017,36(2):369−375. doi: 10.11654/jaes.2016-1237
[5] LEI J, HU X L, YIN D Q, et al. Occurrence, distribution and seasonal variation of antibiotics in the Huangpu River, Shanghai , China[J]. Chemosphere, 2011, 82(6):822−828.
[6] Commossion Regulation No. 508/99 (1999). Official journal of European Community[Z]. 139 March, No. L60.
[7] GOTO T, ITO Y, YAMADA S, et al. High-throughput analysis of tetracycline and penicillin antibiotics in animal tissues using electrospray tandem mass spectrometry with selected reaction monitoring transition[J]. Journal of Chromatography A,2005,1100:193−19. doi: 10.1016/j.chroma.2005.09.056
[8] ORLANDO E A, SIMIONATO A V. Extraction of tetracycline antibiotic residues from fish filet:Comparison and optimization of different procedures using liquid chromatography with fluorescence detection[J]. Journal of Chromatography A,2013,1307:111−118. doi: 10.1016/j.chroma.2013.07.084
[9] GB 31650-2019 食品安全国家标准 食品中兽药最大残留限量[S]. 北京:中国农业出版社, 2020 GB 31650-2019 National food safety standard maximum residue limits of veterinary drugs in foods [S]. Beijing:China Agriculture Press. 2020.
[10] 张启华, 张志军, 田华. 蜂蜜中四环素族抗生素残留量的薄层色谱测定法[J]. 分析测试学报, 1998, 17(4):54−56 ZHANG Q H, ZHANG Z J, TIAN H. Determination of the residues of tetracyclines antibiotics in honey by thin-layer chromatofraphy[J]. Journal of Instrumental Analysis, 1998, 17 (4):54−56.
[11] 张琰图, 章竹君, 孙永华. 高效液相色谱化学发光法检测牛奶中残留四环素类化合物的研究[J]. 化学学报,2006,64(24):2461−2466 doi: 10.3321/j.issn:0567-7351.2006.24.013 ZHANG Y T, ZHANG Z J, SUN Y H. Determination of tetracyclines residues in milk using high-performance liquid chromatography with chemiluminescence detection[J]. Acta Chimical Sinca,2006,64(24):2461−2466. doi: 10.3321/j.issn:0567-7351.2006.24.013
[12] CHEN Y, KONG D Z, LIU L Q, et al. Development of an ELISA and immunochromatographic assay for tetracycline, oxytetracycline, and chlortetracycline residues in milk and honey based on the class-specific monoclonal antibody[J]. Food Anal Methods,2016,9:905−914. doi: 10.1007/s12161-015-0262-z
[13] 檀尊社, 陆恒, 邵伟, 等. 胶体金免疫层析法快速检测水产品中四环素类药物残留[J]. 西北农业学报,2010,19(8):32−37 doi: 10.3969/j.issn.1004-1389.2010.08.007 TAN Z S, LU H, SHAO W, et al. Nanocolloidal gold-based immunoassay for rapid detection of tetracycline residue in aquatic products[J]. Acta Agriculture Boreali-occidentalis Sinica,2010,19(8):32−37. doi: 10.3969/j.issn.1004-1389.2010.08.007
[14] 刘丽, 蔡志斌, 张英. 高效液相色谱法测定水产品中土霉素、四环素、金霉素[J]. 中国卫生检验杂志,2007,17(8):1405−1406 doi: 10.3969/j.issn.1004-8685.2007.08.077 LIU L, CAI Z B, ZHANG Y. Detection of three antibiotics in aquatic products by HPLC[J]. Chinese Journal of Health Laboratory Technology,2007,17(8):1045−1046. doi: 10.3969/j.issn.1004-8685.2007.08.077
[15] 刘艳华, 张纯萍, 门立强, 等. 液相色谱-串联质谱法测定鸡肌肉组织中的四环素类药物残留[J]. 色谱,2006,24(2):171−173 doi: 10.3321/j.issn:1000-8713.2006.02.015 LIU Y H, ZHANG C P, MEN L Q, et al. Determination of tetracycline antibiotics residues in chicken muscle by liquid chromatography-tandem mass spectrometry[J]. Chinese Journal of Chromatography,2006,24(2):171−173. doi: 10.3321/j.issn:1000-8713.2006.02.015
[16] ABBASI M M, NEMATI M, BABAEI H, et al. Solid-phase extraction and simultaneous determination of tetracycline residues in edible cattle tissues using an HPLC-FL method[J]. Iranian Journal of Pharmaceutical Research, 2012, 11(3):781−787.
[17] WANG G N, ZHANG L, SONG Y P, et al. Application of molecularly imprinted polymer-based matrix solid-phase dispersion for determination of fluoroquinolones, tetracyclines, and sulfonamides in meat[J]. Journal of Chromatography B,2017,1065−1066:104−111. doi: 10.1016/j.jchromb.2017.09.034
[18] ZHOU J H, XUE X F, LI Y, et al. Multiresidue determination of tetracycline antibiotics in propolis by using HPLC-UV detection with ultrasonic-assisted extraction and two-step solid phase extraction[J]. Food Chemistry,2009,115:1074−1080. doi: 10.1016/j.foodchem.2008.12.031
[19] LIU Y, YANG H L, YANG S, et al. High-performance liquid chromatography using pressurized liquid extraction for the determination of seven tetracyclines in egg, fish, and shrimp[J]. Journal of Chromatography B,2013(917-918):11−17.
[20] CRISTINABLASCO, DICORCIA A, YOLANDAPICO. Determination of tetracyclines in multi-specie animal tissues by pressurized liquid extraction and liquid chromatography-tandem mass spectrometry[J]. Food Chemistry,2009,116:1005−1012. doi: 10.1016/j.foodchem.2009.03.055
[21] ALBERO B, SANCHEZ-BRUNETE C, GARCIA-VALCARCEL A I, et al. Ultrasound-assisted extraction of emerging contaminants from environmental samples[J]. Trends in Analytical Chemistry,2015,71:110−118. doi: 10.1016/j.trac.2015.03.015
[22] CHEMAT F, ROMBAUT N, SICAIRE A, et al. Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols, and applications. A review[J]. Ultrasonics Sonochemistry,2017,34:540−560. doi: 10.1016/j.ultsonch.2016.06.035
[23] 王 飞, 宓捷波, 李淑静, 等. 改良的QuEChERS 样本前处理/高效液相色谱-串联质谱法检测猪肉中四环素类兽药的残留[J]. 分析测试学报,2017,2(36):272−275 WANG F, MI J B, LI S J, et al. Determination of tetracyclines residues in pork by high-performance liquid chromatography-tandem mass spectrometry with modified QuEChERS sample pretreatment[J]. Journal of Instrumental Analysis,2017,2(36):272−275.
[24] GRANDE-MARTINEZA M, MORENO-GONZALEZB D, ARREBOLA-LIEBANASA F J, et al. Optimization of a modified QuEChERS method for the determination of tetracyclines in fish muscle by UHPLC-MS/MS[J]. Journal of Pharmaceutical and Biomedical Analysis,2018,155:27−32. doi: 10.1016/j.jpba.2018.03.029
[25] TOLGYESI A, BEKESI L T, FEKETE V S. Determination of tetracyclines in pig and other meat samples using liquid chromatography coupled with diode array and tandem mass spectrometric detectors[J]. Meat Science,2014,96:1332−1339. doi: 10.1016/j.meatsci.2013.11.011
[26] MARAN J P, MANIKANDAN S, THIRUGNANASAMBANDHAM K, et al. Box-Behnken design-based statistical modeling for ultrasound-assisted extraction of corn silk polysaccharide[J]. Carbohydrate Polymers,2013,92 (1):604−611. doi: 10.1016/j.carbpol.2012.09.020
[27] CURRIE L, HORWITZ W. IUPAC recommendations for defining and measuring detection and quantification limits[J]. Anal Magazine. 1994, 22:M24–26.
[28] CHATTERJEE N S, UTTARA S, BANERJEE K, et al. Multiresidue analysis of multiclass pesticides and polyaromatic hydrocarbons in fatty fish by gas chromatography-tandem mass spectrometry and evaluation of matrix effect[J]. Food Chemistry,2016,196:1−8. doi: 10.1016/j.foodchem.2015.09.014
[29] DIAO P P, CHEN Q, WANG R, et al. Phenolic endocrine-disrupting compounds in the Pearl River Estuary:Occurrence, bioaccumulation and risk assessment[J]. Sci Total Environ,2017,584−585:1100−1107. doi: 10.1016/j.scitotenv.2017.01.169
[30] RAMASWAMY B R, KIM J, ISOBE T, et al. Determination of preservative and antimicrobial compounds in fish from Manila Bay, the Philippines using ultra-high performance liquid chromatography-tandem mass spectrometry, and assessment of human dietary exposure[J]. Journal of Hazardous Materials,2011,192:1739−1745. doi: 10.1016/j.jhazmat.2011.07.006
[31] OMAR T F T, ARIS A Z, . YUSOFF F M, et al. Occurrence and level of emerging organic contaminant in fish and mollusk from Klang River estuary, Malaysia and assessment on human health risk[J]. Environmental Pollution,2019,248:763−773. doi: 10.1016/j.envpol.2019.02.060
[32] LI R J, LIU T J, CUI S H, et al. Residue behaviors and dietary risk assessment of dinotefuran and its metabolites in Oryza sativa by a new HPLC-MS/MS method[J]. Food Chemistry,2017,235:188−193. doi: 10.1016/j.foodchem.2017.04.181
[33] CAARASCO-PANCORBO A, CASADO-TERRONES S, SEGURA-CARRETERO A, et al. Reversed-phase high-performance liquid chromatography coupled to ultraviolet and electrospray time-of-flight mass spectrometry on-line detection for the separation of eight tetracyclines in honey samples[J]. Journal of Chromatography A,2008,1195:107−116. doi: 10.1016/j.chroma.2008.05.003
[34] CRISTOFANI E, CHIARA ANTONINI, TOVO G, et al. A confirmatory method for the determination of tetracyclines in muscle using high-performance liquid chromatography with diode-array detection[J]. Analytica Chimica Acta,2009,637:40−46. doi: 10.1016/j.aca.2008.10.029
[35] WALSH J R, WALKER L V, WEBBER J J. Determination of tetracyclines in bovine and porcine muscle by high-performance liquid chromatography using solid-phase extraction[J]. Journal of Chromatography A,1992,596(2):211−216. doi: 10.1016/0021-9673(92)85009-I
[36] OKA H, ITO Y, MATSUMOTO H. Chromatographic analysis of tetracycline antibiotics in foods[J]. Journal of Chromatography A,2000,882(1-2):109−113. doi: 10.1016/S0021-9673(99)01316-3
[37] WEI Y M, ZHANG Y, XU J, et al. Simultaneous quantification of several classes of antibiotics in water, sediments, and fish muscles by liquid chromatography-tandem mass spectrometry[J]. Frontiers of Environmental Science & Engineering,2014,8:357–371.
[38] VARGASMAMANI M C, AMAYAFARFAN J, REYESREYES F G, et al. Simultaneous determination of tetracyclines in pharmaceuticals by CZE using experimental design[J]. Talanta,2006,70(2):236−243. doi: 10.1016/j.talanta.2006.02.048
[39] MELO A, AGUIAR A, MANSILHA C, et al. Optimisation of a solid-phase microextraction/HPLC/Diode Array method for multiple pesticide screening in lettuce[J]. Food Chemistry,2012,130:1090–1097. doi: 10.1016/j.foodchem.2011.07.137
[40] MILLER J N, MILLER J C. Statistics and chemometrics for analytical chemistry[M]. fifth, Pearson Education Limited, England, 2005.
[41] CARLOS P V, STEPHEN L, TREVOR W, et al. Mixture-amount design and response surface modeling to assess the effects of flavonoids and phenolic acids on developmental performance of Anastrepha ludens[J]. Journal of Chemical Ecology,2014,40(3):297−306. doi: 10.1007/s10886-014-0404-6
[42] MONTEIRO S H, FRANCISCO J G, CAMPION T F, et al. Multiresidue antimicrobial determinationin Nile tilapia ( Oreochromis niloticus) cage farming by liquid chromatographytandem mass spectrometry[J]. Aquaculture,2015,447:37–43. doi: 10.1016/j.aquaculture.2015.07.002
[43] PEREIRA-LOPES R, CAZORLA-REYES R, ROMERO-GONZALEA R, et al. Multiresidue determination of veterinary drugs inaquaculture fish samples by ultra high performance liquid chromatographycoupled to tandem mass spectrometry[J]. J. Chromatogr. B , 2012,895:39–47.
-
期刊类型引用(1)
1. 白俊露,曾军杰,何鹏飞,李佩佩. QuEChERS-超高效液相色谱串联质谱法同时检测水产品中11种四环素类药物. 浙江海洋大学学报(自然科学版). 2024(04): 335-345 .  百度学术
百度学术
其他类型引用(2)
 下载:
下载:





计量
- 文章访问数: 71
- HTML全文浏览量: 9
- PDF下载量: 8
- 被引次数: 3



